UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
For the fiscal year ended
or
For the transition period from _____ to _____
COMMISSION FILE NUMBER:
(Exact name of registrant as specified in its charter)
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
(Address of principal executive offices) (Zip Code)
Registrant’s telephone number, including area code
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading symbol | Name of exchange on which registered | ||
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a
well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not
required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant:
(1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months
(or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements
for the past 90 days.
Indicate by check mark whether the registrant
has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (Section 232.405
of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| ☒ | Smaller reporting company | ||
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant
has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial
reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or
issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant
is a shell company (as defined in Rule 12b-2 of the act): Yes ☐ No
The aggregate market value of voting stock held
by nonaffiliates of the registrant as of June 30, 2022, the last business day of the registrant’s most recently completed second
fiscal quarter, based on the closing price of the common stock on the NYSE American on June 30, 2022 was $
As of March 31, 2023,
Table of Contents
i
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (this “Report”) contains forward-looking statements that involve risks and uncertainties, principally in the sections entitled “Description of Business,” “Risk Factors,” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” All statements other than statements of historical fact contained in this Report, including statements regarding future events, our future financial performance, business strategy and plans and objectives of management for future operations, are forward-looking statements. We have attempted to identify forward-looking statements by terminology including “anticipates,” “believes,” “can,” “continue,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “should,” or “will” or the negative of these terms or other comparable terminology. Although we do not make forward-looking statements unless we believe we have a reasonable basis for doing so, we cannot guarantee their accuracy. These statements are only predictions and involve known and unknown risks, uncertainties and other factors, including the risks outlined under “Risk Factors” or elsewhere in this Report, which may cause our or our industry’s actual results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time and it is not possible for us to predict all risk factors, nor can we address the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause our actual results to differ materially from those contained in any forward-looking statements. All forward-looking statements included in this document are based on information available to us on the date hereof, and we assume no obligation to update any such forward-looking statements.
You should not place undue reliance on any forward-looking statement, each of which applies only as of the date of this Report. Before you invest in our securities, you should be aware that the occurrence of the events described in the section entitled “Risk Factors” and elsewhere in this Report could negatively affect our business, operating results, financial condition and stock price. Except as required by law, we undertake no obligation to update or revise publicly any of the forward-looking statements after the date of this Report to conform our statements to actual results or changed expectations.
ii
PART I
ITEM 1. BUSINESS.
Description of Our Business
Actinium Pharmaceuticals, Inc. (“Actinium”) is a biopharmaceutical company developing targeted radiotherapies to deliver cancer-killing radiation with cellular level precision to treat patients with high unmet medical needs. Our vision is to build a specialty, hospital focused radiotherapeutics company that develops and markets medicines for relapsed or refractory cancer patients who are treated primarily in large quaternary care hospitals and their catchment areas. We intend to leverage the clinical data of our lead product candidates, Iomab-B and Actimab-A, to improve outcomes in patients with relapsed or refractory acute myeloid leukemia (“r/r AML”) by launching two radiotherapy drugs in 5 years that address the significant need for better outcomes from treatment with therapeutics or from undergoing a bone marrow transplant (“BMT”).
We also intend during this time frame to further advance Iomab-B outside of acute myeloid leukemia (“AML”) based on promising data as a disease control and conditioning agent for various other blood cancers. Based on early promising clinical trial results, we are also working on a lower dose next generation conditioning program, Iomab-ACT, for rapidly growing cell and gene therapies.
Our Clinical Pipeline
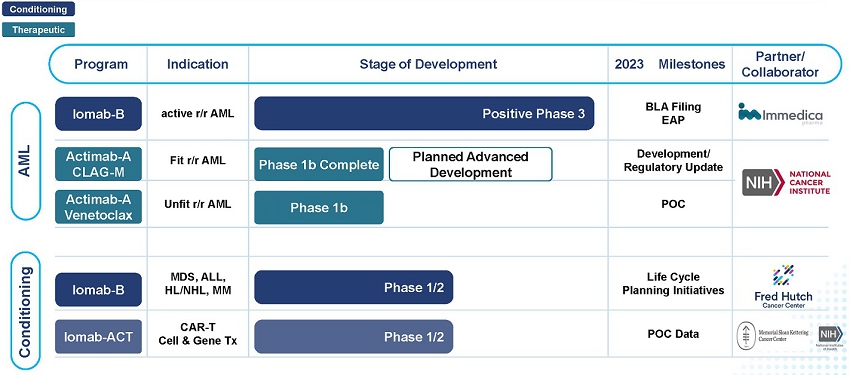
AML is an aggressive, heterogeneous disease that is difficult-to-treat. Most AML patients develop relapsed or refractory disease within one year and have an extremely poor prognosis and dismal survival. Currently, a BMT is the only curative regimen available for AML patients, however, access is limited to AML patients who are fit enough to withstand the challenges associated with this treatment. The majority of AML patients are considered not transplantable in routine clinical practice as they are not fit enough to withstand the rigors of the patient journey which includes: therapy to attain a remission, conditioning regimens to destroy diseased marrow, challenge of the transplant itself or post-transplant complications.
Our Iomab-B and Actimab-A product candidates fill the major unmet medical needs in r/r AML in a complementary fashion as they are directed at different parts of the patient journey. Iomab-B is a targeted bridging therapy that provides both disease control and conditioning in one agent. Results from a phase 3 trial has demonstrated unprecedented access to a BMT and improved survival in unfit patients who are currently not considered transplantable in routine clinical practice. Actimab-A is a targeted therapy for fit patients that has demonstrated an impressive extension in survival in a proof-of-concept study and is poised for advanced development in collaboration with the NCI, or National Cancer Institute. Together, they provide us the opportunity to transform the treatment of AML, especially in the relapsed and refractory segment which represents over 50% of AML patients.
1
On October 31, 2022, we announced topline results from the pivotal Phase 3 Study of Iomab-B in Elderly Relapsed or Refractory AML or SIERRA trial, which demonstrated that Iomab-B met the primary endpoint of durable Complete Remission (“dCR”) with a high degree of statistical significance (p<0.0001). On February 18, 2023, we announced full trial results, demonstrating unprecedented access, improved outcomes and better safety and tolerability with double 1-year and median overall survival (“OS”) compared to control arm patients receiving Iomab-B at the 2023 Tandem Meetings aka the Transplantation & Cellular Therapy Meetings of the American Society for Transplantation and Cellular Therapy (“ASTCT”) and the Center for International Blood & Marrow Transplant Research (“CIBMTR”). We believe these results provide the opportunity to establish Iomab-B as a new standard of care, and if approved, we intend to commercialize the product in the United States (“U.S.”).
On April 12, 2022, we announced a commercialization agreement for Iomab-B with Immedica AB (“Immedica”) for exclusive rights in Europe, the Middle East and North Africa (“EUMENA”). Actinium received an upfront payment of $35 million USD with the potential for an additional $417 million USD in regulatory and sales milestones and mid-twenty percent royalties. The market in the EU region is attractive due to the higher incidence of AML and number of BMT procedures compared to the U.S. with the same unmet patient need, and our partnership with Immedica positions us well to capitalize on this opportunity.
Actimab-A is the industry-leading program, leveraging the potent alpha radiation emitting isotope Actinium-225 (“Ac-225”) based on clinical development in approximately 150 patients treated over 6 clinical trials. The potent linear energy transfer emitted by Ac-225 has no known resistance mechanism. Actimab-A is being developed in combination with other regimens to exploit mechanistic synergies and leverage the mutation-agnostic mechanism of action of Ac-225 with the objective of establishing it as a backbone therapy in AML, an extremely heterogenous disease.
On December 10, 2022, we shared the Phase 1 results from the Actimab-A CLAG-M combination trial at the American Society of Hematology (“ASH”) Annual Meeting & Exposition that showed high response rates and minimal residual disease (“MRD”) negativity, translating to a meaningful survival benefit of 53%and 32%at one and two years in patients who are typically expected to live two to four months. At the same meeting, we shared Phase 1 data showing that the combination of Actimab-A + venetoclax was well-tolerated with responses, including a Complete Remission (“CR”) and a partial response in early dose escalation cohorts. We believe the promise of these results paved the way for the NCI Cooperative Research and Development Agreement (“CRADA”), announced on February 6, 2023, to develop Actimab-A for the treatment of patients with AML and other hematologic malignancies.
Our differentiated R&D is further exemplified by our next-generation Iomab-ACT conditioning program for rapidly growing cell and gene therapies, as well as our solid tumor and immunotherapy collaborations with Astellas Pharma Inc. (“Astellas”), AVEO Oncology/LG Chem (“LG Chem”) and EpicentRx, Inc. (“EpicentRx”). In addition, we have several other programs in solid tumors at the pre-clinical stage with investigational new drug (“IND”) enabling studies ongoing and our extensive intellectual property (“IP”) portfolio includes over 200 issued patents and pending patent applications worldwide.
We are actively working on launching an early access program (“EAP”) for Iomab-B and intend to file a Biologics License Application (“BLA”) by year-end while preparing for a U.S. commercial launch and working with our partner Immedica to support the Marketing Authorization Application (“MAA”) and commercialization in the EU. Late-stage Actimab-A development is expected to begin in the second half of 2023 under the NCI CRADA and is anticipated to have a material balance sheet sparing impact over the next several years. We expect to create significant value due to the combination of major milestones and balance sheet strength with the approximately $100 million cash on hand at year-end 2022 projected to fund operations through 2025 as we continue to drive ahead with realizing our five-year plan.
Market Opportunity
The market opportunity for Iomab-B and Actimab-A, as depicted in the diagram below, exists in AML and for cellular therapy conditioning in various blood cancers. We believe that Iomab-B and Actimab-A can fill the major unmet medical needs in r/r AML in a complementary fashion as they are utilized in different parts of the patient treatment journey. The incidence of AML is approximately 21,000 patients per year, with a prevalence of approximately 70,000 in the U.S., (approximately 27,500 new patients per year in Europe) and the disease has an outsized economic impact relative to its population size. Over 50% of patients diagnosed with AML will develop relapsed or refractory disease, with a median age of 68 years at diagnosis. Despite 10 new approved therapies since 2017, no significant advancements have been made toward a cure and there is a significant unmet need for better therapeutics, which provide the opportunity for Actimab-A. Actimab-A is a targeted radiotherapy for fit patients that has demonstrated an impressive improvement in survival in a proof-of-concept study and is poised for advanced development in collaboration with the NCI. Using Actimab-A in combination with chemotherapy or a targeted therapy, we have the potential opportunity to treat both newly diagnosed or r/r AML patients, with the potential addressable population comparable to the prevalence of patients with AML.
2
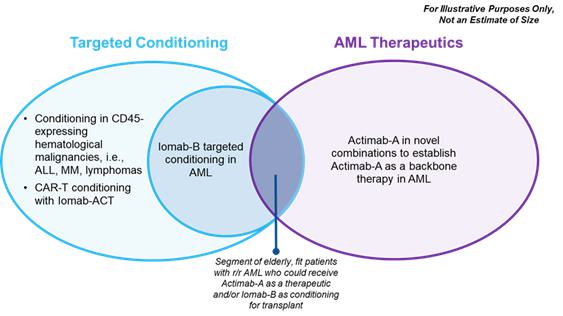
Today, less than 20% of AML patients are able to access a BMT, currently the only potentially curative option. These patients are usually younger, fit, and able to withstand the challenges associated with this treatment, leaving the large majority of AML patients ineligible for transplant. This provides an opportunity for Iomab-B, which has demonstrated the ability to enable unfit patients to benefit from a BMT. Thus Iomab-B can potentially expand the market from the approximately 400 r/r AML patients who are transplanted currently to approximately 8,000 unfit patients that could be eligible for transplant. Iomab-B has demonstrated the ability to improve BMT access with extended survival and potentially curative outcomes in several other hematological diseases outside of AML. Several clinical trials in over 300 patients with myelodysplastic syndromes (“MDS”), acute lymphocytic leukemia (“ALL”), Hodgkin’s lymphoma (“HL”), Non-Hodgkin lymphoma (“NHL”) and multiple myeloma (“MM”) have demonstrated the same value proposition as in AML. This data provides an opportunity to expand the market for Iomab-B beyond AML via label expansion. In the U.S., there are approximately 185,000 patients diagnosed with blood cancers (e.g., leukemia, lymphoma, and myeloma) that are treatable with BMT, of which, only approximately 20,000 are transplanted, leaving greater than 165,000 patients who could potentially benefit from transplant. These patients do not receive a BMT today primarily because they are unfit with active disease and are not considered eligible, as they cannot tolerate the rigors of therapy required to induce a remission and the conditioning agents required to ablate the marrow prior to a BMT.
Beyond BMT, the opportunity exists for better conditioning in other areas of cellular therapy such as CAR-T as well as gene therapies. The pipeline of CAR-T and gene therapies has rapidly expanded, with the addressable patient population expected to nearly double in the next one to five years and reach approximately 93,000 patients in the U.S. by 2030 based on the current pipeline. The CAR-T market size in terms of dollars is estimated to grow at a CAGR of approximately 11% over the next 5 plus years. The addressable market for Iomab-ACT is in line with the patient population for cellular therapy as all patients receive conditioning of some type prior to these treatments. We will continue to develop Iomab-ACT, our lower dose, next generation conditioning program for rapidly growing cell and gene therapies based on early promising results, ultimately with the value proposition of improving overall access and outcomes for patients who need cellular or gene therapies.
Our Strategy
Actinium’s strategy is to build a fully-integrated, specialty radiotherapeutics company focused on the top 100 cancer hospitals using the power of our platform to deliver new treatment options for patient populations living with high unmet medical needs in hematology and oncology. We believe our focus on relapsed and refractory disease in cancer indications with high unmet medical need, with limited or no competition, and where the primary delivery of care occurs in a few large comprehensive cancer care centers, is the appropriate strategy for our company. The cell killing power of linear energy transfer delivered via radiotherapeutics is unmatched by other technologies and we believe relapsed, refractory disease is an area where radiotherapeutics can succeed over other approaches. However, radiotherapeutics must be delivered on a just-in-time basis, and commercial and supply chain barriers are higher than with other types of medicines. The validity of our approach is demonstrated by our product development strategy as well as the commercial and operating model that we are building for our lead product candidates, Iomab-B and Actimab-A.
3
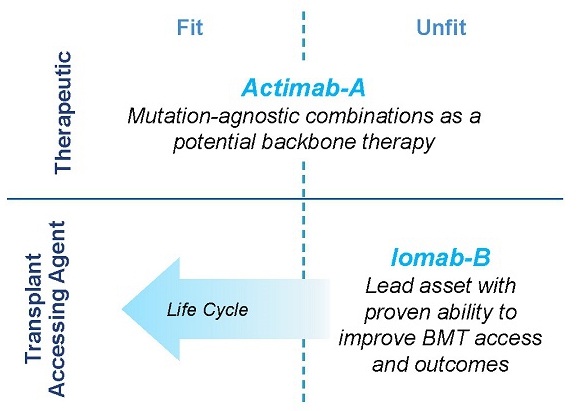
We intend to transform the treatment of AML with our Iomab-B and Actimab-A product candidates, each of which has demonstrated impressive extension of survival in the most difficult-to-treat patients who are typically expected to survive two to four months. The r/r AML segment comprises over 50% of all AML patients. Actimab-A, a therapeutic agent, and Iomab-B for induction and conditioning, can be used in a complementary fashion as depicted in the diagram below. Based on solid clinical evidence with these product candidates, we intend to develop and commercialize these two radiotherapy drugs, starting with Iomab-B in 2024 and Actimab-A in 2027, if approved, to improve survival in patients with r/r AML.
Iomab-B and Actimab-A have the potential to significantly improve r/r AML outcomes in a complementary manner
The operating model required to achieve our vision is attractive for several reasons, including the concentrated point of care; the top 50 transplant centers account for approximately 75% of BMTs and the top 100 hospitals treat over 50 percent of r/r AML patients. Further, there is significant overlap in the healthcare providers and ecosystem required to diagnose, treat and care for r/r AML patients within these hospitals, which will enable us to deploy a relatively small commercial organization and operate a supply chain without the need for large investments. Our product pipeline is targeting a broader opportunity in conditioning via label expansion of Iomab-B into BMT for other blood cancers and with Iomab-ACT, our next generation conditioning program for rapidly growing cell and gene therapies. Further, our solid tumor programs are initially directed at relapsed or refractory cancers, a stage of disease where treatment is again concentrated in large hospitals, which account for a significant portion of patients. We believe our strategy will enable us to build a successful company with high operating efficiencies and is feasible to achieve without requiring a commercial partner.
Our strategic priorities are to:
| ● | Establish Iomab-B as the standard of care to improve BMT access and survival outcomes in r/r AML patients who are currently not considered transplantable in routine clinical practice: We intend to file a BLA in the second half of 2023 based on the strong results from the Pivotal Phase 3 SIERRA trial and leverage our proven operating track record at key cancer centers to build a high impact organization that can effectively commercialize Iomab-B. By virtue of the SIERRA trial, we have established operations at 24 leading BMT centers that represent about 30% of transplant volume in the U.S. and have strong working partnership with Key Opinion Leaders and their teams. The positive SIERRA results demonstrating unprecedented access to BMT and outcomes along with our commitment to operational excellence provides a strong foundation for our commercial team in the U.S. We will also work with our partner Immedica to file the MAA for the EU and support Iomab-B’s potential approval and launch with our expertise, as well as supply drug product for commercialization. |
4
| ● | Advance Actimab-A in combinations as a backbone therapy for r/r AML: We intend to progress late-stage development of Actimab-A to leverage its mutation-agnostic mechanism of action and exploit synergies by combining with other treatments to develop it as an AML backbone therapy. This approach is validated by proof-of-concept data from our Actimab-A + CLAG-M combination trial in r/r AML, which included 57% of patients who had failed venetoclax and live two to four months on average. The results demonstrated high response rates overall and in these venetoclax failed patients median overall survival was 59% at one year and thirty-two percent at two years. Our collaboration with the NCI under the CRADA will provide broad support for late-stage development of Actimab-A + CLAG-M and also other clinical trials to broaden use while preserving our balance sheet. Actimab-A, if approved, would enable us to launch a second product that is complementary to Iomab-B and fulfill our ambition of radically transforming the treatment outcomes of r/r AML and expand our commercial footprint into the remaining top 100 cancer care centers outside of the leading BMT hospitals. |
| ● | Expand the Iomab-B label and revenue stream via life cycle management: We intend to leverage data from several clinical trials that demonstrate the ability of Iomab-B to improve BMT access and outcomes in five additional hematologic indications. These data in MDS, ALL, HL, NHL and MM provide the foundation to expand the label for Iomab-B and increase its market potential. In AML, we would seek label expansion into haploidentical transplants, earlier lines of treatment and younger patients below the age of 55, the cutoff in the SIERRA trial. As much as possible, we would seek to use investigator sponsored trials as the primary strategy for label expansion in order to maximize capital utilization. |
| ● | Further expand our conditioning franchise by developing Iomab-ACT for cell and gene therapies: We plan to develop Iomab-ACT to be used for either lymphodepletion or reduced intensity conditioning prior to CAR-T and gene therapies. Similar to BMT, access and outcomes of patients who might benefit from these therapies is limited by sub-optimal chemotherapy-based conditioning agents. The number of patients potentially eligible for Iomab-ACT is growing with increased availability of commercial cell and gene therapy products, as well as the expanding number of indications. We are studying Iomab-ACT in conditioning prior to CAR-T cellular therapy via a National Institutes of Health (“NIH”) funded clinical trial with Memorial Sloan Kettering Cancer Center (“MSKCC”). We expect to present proof-of-concept data from this study in the second half of 2023 and announce further development of this program in the CAR-T space. |
| ● | Leverage our R&D capabilities and technological prowess to advance our solid tumor programs and partnerships: We intend to continue to direct our R&D effort to advance our solid tumor programs into the clinic and support life cycle management for Iomab-B and Actimab-A. Our solid tumor programs and technological capabilities are validated by our partnerships with Astellas, LG Chem, and EpicentRx, which are focused on solid tumors and immunotherapies. Our R&D prowess is demonstrated by our robust patent portfolio with over 200 issued and pending patent applications worldwide which include protection for Iomab-B into 2037. Our IP portfolio also includes several patent families to manufacture Ac-225 in a cyclotron, and valuable know-how. |
In keeping with our strategic vision over the next five years, we would first focus our energies on ensuring an Iomab-B approval and successful launch into core BMT centers to ensure commercial success. We intend to expand the Iomab-B label and revenue stream in a capital efficient manner while progressing the development of Actimab-A by leveraging the NCI CRADA and its balance sheet sparing opportunity. We will progress the development of Iomab-ACT to proof-of-concept and explore partnerships as a means to achieve commercialization. Our solid tumor programs will progress toward the clinic as we continue to build out our commercial footprint into the top 100 hospitals leaving us well-positioned to develop them in accordance with our vision. With commercial dynamics aligning favorably for a successful Iomab-B launch and with late-stage development of Actimab-A in collaboration with the NCI, we plan to deliver on our mission to transform the treatment of AML and patient outcomes, and create a highly differentiated, specialty radiotherapeutics company focused on the top 100 large hospitals.
5
Our Product Pipeline
We have strategically focused our development efforts in areas where there is a significant unmet medical need. We are developing a portfolio of novel radiotherapeutics that has the potential to positively impact the outcomes of people living with hard-to-treat diseases such as r/r AML via both a therapeutic and induction/conditioning agent. Outside of AML, our pipeline development offers the opportunity to enhance the value proposition of cell and gene therapies with our targeted conditioning programs.
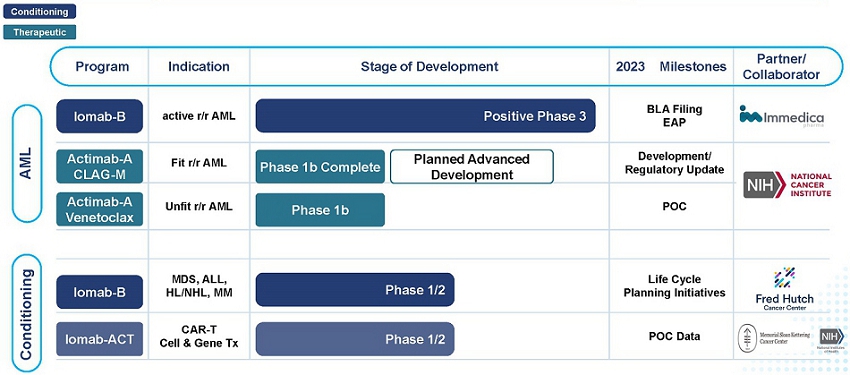
AML Focused Programs – Iomab-B and Actimab-A
Our Iomab-B and Actimab-A product candidates are focused on addressing the major unmet medical needs in r/r AML in a complementary manner and are directed at different parts of the patient journey.
Iomab-B – Targeted Radiotherapeutic for Induction and Conditioning. A potential new standard of care enabling a curative BMT in currently non-transplantable r/r AML patients with poor survival prognosis
Opportunity to Change the Current Paradigm for Accessing a BMT and Improving Outcomes
The current approach in preparing patients for a BMT is to first induce a remission with therapeutic agents to reduce the disease burden and then suppress or destroy the patient’s immune system, including the diseased bone marrow, with conditioning regimens prior to transplanting the healthy donor hematopoietic stem cells, which are expected to restore normal bone marrow function following engraftment. As this approach requires patients to withstand multiple challenges from non-targeted therapies, which include chemotherapy agents and/or total body irradiation that are highly toxic, BMT is typically limited to FIT patients. Iomab-B is a targeted therapy that provides both disease control and conditioning in one agent and is well-tolerated even by UNFIT patients who typically are not transplanted in routine practice today. The SIERRA trial was designed to demonstrate that UNFIT patients with active disease could be administered Iomab-B and proceed directly to a BMT without the need for inducing a remission and that this approach could result in improved survival and curative outcomes. As seen by the positive results of the SIERRA trial detailed below, Iomab-B represents an exciting new paradigm in the management of AML patients and establishes a potential new standard of care especially for UNFIT patients in the relapsed or refractory setting.
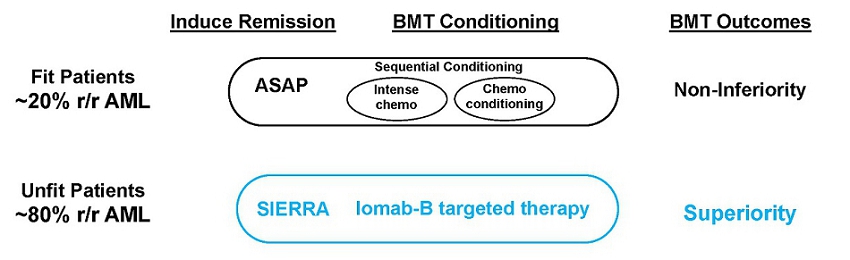
A similar approach has also been tried in the Phase 3 ASAP trial but with FIT patients. However, to avoid confusion between the potential of the approaches used in the ASAP and SIERRA trials, important distinctions between these trials are depicted in the graphic below. The ASAP trial sought to demonstrate non-inferiority between two non-novel approaches and found that outcomes similar to those of current practice could be achieved without first getting a patient into remission before taking them to BMT by giving them sequential conditioning or treating them twice with non-targeted chemotherapy agents that are typically used in this setting.
The ASAP approach is limited to only FIT patients as the UNFIT patients treated in SIERRA could not tolerate ASAP’s highly toxic sequential conditioning approach. Sequential conditioning is not novel as a similar trial to ASAP was conducted by the UK National Cancer Research Institute in 2019, which did not show any benefit from this approach in high-risk AML and MDS patients (Craddock et al. Augmented Reduced Intensity Regimen Does Not Improve Postallogeneic Transplant Outcomes in Acute Myeloid Leukemia. J Clin Oncol. 2021). The SIERRA trial results therefore can change the paradigm in transplant because non-transplantable patients in routine clinical practice can benefit from a transplant with Iomab-B and have superior outcomes. While both approaches in these trials support increased access to BMT, only Iomab-B is applicable to the unfit patients who comprise approximately 80%of r/r AML patients and can potentially expand the market for transplant.
6
Schetelig et al. Results from the Randomized Phase III ASAP Trial. ASH 2022
Pivotal Phase 3 SIERRA Trial for Iomab-B (131Iodine-apamistamab)
The SIERRA trial was designed to demonstrate the ability of Iomab-B to overcome challenges related to patient access to curative BMT. Unfortunately, approximately 30% of patients with AML have primary refractory disease while 50% relapse quickly after achieving initial remission. Getting these patients with primary r/r AML into remission is very challenging due to characteristics such as age, comorbidities, and disease features such as high-risk mutations that contribute to lack of response to salvage therapies and limit treatment options.
Patients must be able to overcome several challenges related to curative BMT. The first access challenge is that the patient needs to be in complete remission prior to BMT. The current clinical practice is not to transplant patients with active AML as outcomes are poor due to high relapse rates. The National Comprehensive Cancer Network (“NCCN”) guidelines also recommend treatment to achieve remission prior to transplant in patients with relapsed AML. The second challenge to access is tolerance to current conditioning regimens. For older patients, myeloablative regimens are not an option due to intense toxicity and mortality. The third challenge is the ability to achieve post-BMT remission and successful engraftment. Inadequate conditioning can lead to graft failure, which is associated with very high mortality. Patients who fail to achieve a CR post-transplant have extremely poor outcomes and a survival of a few weeks. The fourth challenge relates to BMT tolerability and post-BMT complications. The conditioning and immunosuppressive regimens given to these patients put them at high risk for infectious complications and toxicity. In the SIERRA trial, Iomab-B addresses all four of these challenges. Access to BMT is improved as CR is not needed pre-BMT given effective disease control and targeted myeloablation. With better post-BMT engraftment, CR and lower complications, the SIERRA trial also addressed the challenges related to improved outcomes through Iomab-B.
7
The SIERRA results, presented in the late-breaker session at the 2023 Tandem Meetings: Transplantation & Cellular Therapy Meetings of the ASTCT and the CIBMTR, support Iomab-B’s value proposition of enabling both improved access and outcomes of a BMT, thereby providing a significant curative option for r/r patients, a segment that represents approximately 50% of all AML patients and the majority not transplanted today. The design of the SIERRA trial is provided in the figure below.
SIERRA: A Novel, Pivotal Phase 3 Study of Iomab-B in r/r AML
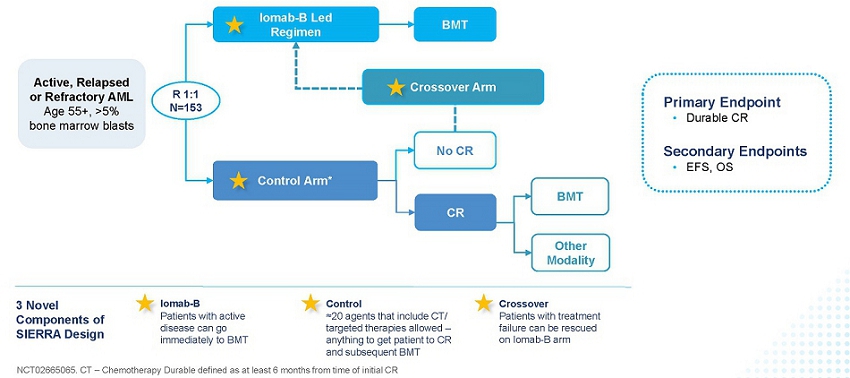
The pivotal Phase 3 SIERRA trial is a 153-patient, randomized, multi-center, controlled trial of Iomab-B in patients aged 55 and above with active r/r AML, who were heavily pre-treated and had high-risk characteristics. Patients enrolled had blast counts of 5% or greater in the marrow or circulating blasts suggestive of active AML. In this study, Iomab-B was compared to the control arm that allowed physician’s choice of over 20 available agents, including chemotherapies and/or targeted therapies such as venetoclax (BCL-2 inhibitor), FLT3 inhibitors, IDH inhibitors and Mylotarg, reflecting current best treatment practices attempting to get patients to CR. The control arm included recently approved AML therapies that were added to the SIERRA protocol as they became available. The crossover arm was designed in SIERRA for an equipoise that offered Iomab-B to patients failing to achieve a CR on the control arm with an intent to rescue them by taking them to transplant. Of note, SIERRA had highly restrictive optionality for post-transplant maintenance. Patients with active, r/r AML are not considered eligible for BMT with current approaches and the SIERRA trial was the only randomized Phase 3 trial to offer BMT as a treatment option for this patient population. These patients would not be offered BMT in standard practice and therefore have dismal survival outcomes of two to three months. The primary endpoint of the SIERRA trial was dCR of 180 days and the secondary endpoints are OS and Event-Free Survival (“EFS”). The comparison of OS in subjects randomized to the control arm who crossed over to receive Iomab-B versus all others in the control group was an exploratory efficacy endpoint.
8
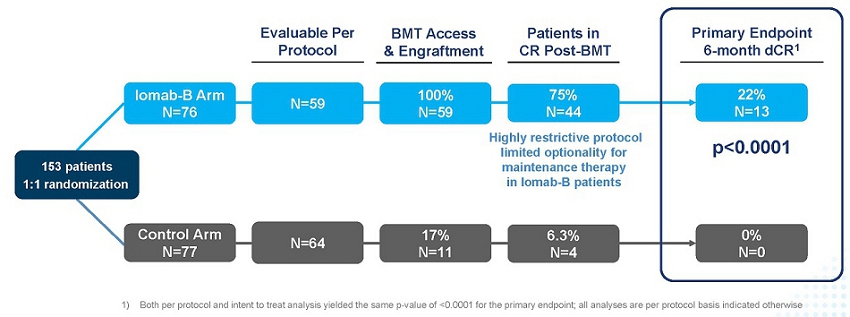
As seen in the graphic below, the primary endpoint of 6-month dCR was met with a high degree of statistical significance (p<0.0001). 75% of patients (44/59) receiving Iomab-B achieved an initial remission 30 days after their BMT compared to 6.3% of patients (4/64) in the control arm. 22% of the patients receiving Iomab-B maintained dCR lasting 180 days or more despite limited optionality for post-transplant maintenance, while none of the patients on the control arm achieved dCR. The current standard practice is to administer post-transplant maintenance therapy to reduce chances of relapse. The results presented below are on a per protocol basis, which means that only data that was in strict adherence to the protocol without any deviations was considered for the analysis. It is important to note that the p-value of the primary endpoint in the intent-to-treat analysis was <0.0001, the same as the per protocol analysis.
SIERRA Results: Iomab-B Meets Primary Endpoint with High Significance (p<0.0001)
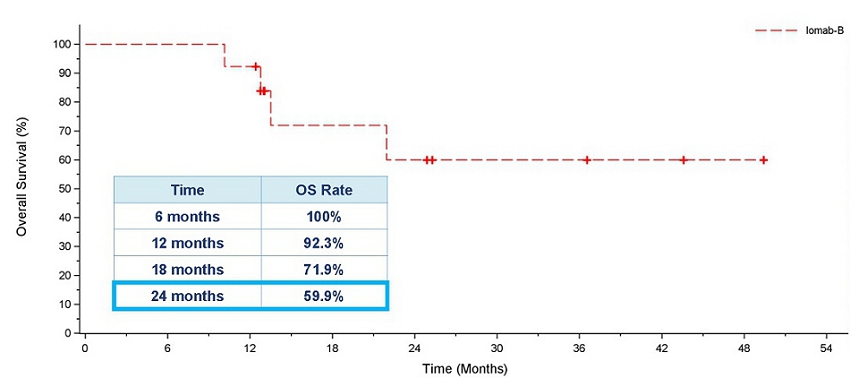
As demonstrated in the OS graph below, patients who achieved 6-month dCR had 92.3% 1-year survival and 59.9% 2-year survival. Median OS had not been reached in these patients. It is worth noting that two years in CR is a significant milestone in this patient population, highly indicative of long-term survival and a possible curative outcome.
9
Overall Survival for Patients who Achieved 6-month dCR with Iomab-B
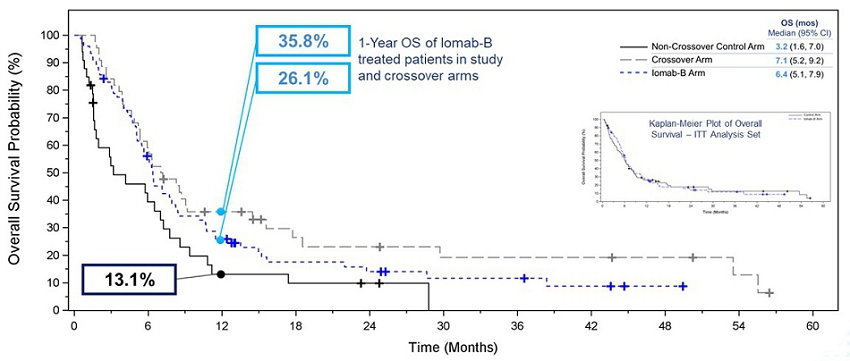
OS was one of the secondary endpoints of the study. The Kaplan-Meier plot in the inset of the graph below shows Intent-to-Treat (“ITT”) OS results between the Iomab-B arm and the control arm. Due to the crossover design, ITT analysis of OS was confounded by the early crossover of patients (within 28 days) from the control arm to the Iomab-B arm (57.1%). The effective rescue of these crossover patients by Iomab-B led to an outsized contribution of the Iomab-B effect on control arm patients. As a result, median OS in the Iomab-B arm was similar to that in the control arm and this secondary endpoint was not met in the ITT analysis.
In order to isolate the true impact of Iomab-B on OS, one of the exploratory efficacy endpoints was the comparison of OS in subjects randomized to the control arm who crossed over to receive Iomab-B versus all others in the control arm, as well as the control arm patients who did not crossover versus the Iomab-B arm. The Kaplan-Meier plot of OS in the graphic below shows that this exploratory analysis demonstrated the clear benefit of Iomab-B over the control arm. The median OS for the Iomab-B group was 6.4 months which was double the 3.2 months for the non-crossover patients in the control arm. Patients who crossed over from the control arm to receive Iomab-B had a median OS of 7.1 months demonstrating further the ability of Iomab-B to treat patients who are non-treatable by conventional means.
A similar pattern favoring the Iomab-B group was seen across the pre-defined subgroups, where 1-year OS for Iomab-B was 26.1% compared with 13.1% for the non-crossover control arm. The 1-year OS for patients in the crossover arm was 35.8%. This clearly demonstrates the OS benefit of Iomab-B over the control arm and two to three-fold improvement in survival outcomes possible with its use.
10
Kaplan-Meier Plot of Overall Survival ‒ Iomab-B, Crossover, and Non-Crossover Control Arm
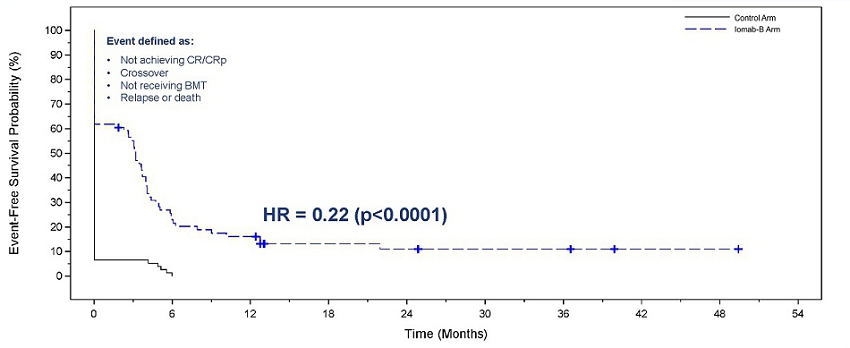
Iomab-B produced a significant and clinically meaningful improvement in the secondary endpoint of EFS, with a 78% reduction in the probability of an event (Hazard Ratio=0.22, p<0.0001 for both per protocol and ITT basis). EFS at 180 days for the Iomab-B arm was 28% compared to 0.2% for the control arm. In the SIERRA trial, an event is defined as one of the following: a patient not achieving CR/CRp or crossing over, patient not receiving BMT, a patient relapsing or death.
In the figure below comparing EFS with Iomab-B versus the control arm, the initial vertical drop in the curve in the Iomab-B arm represents those patients who did not achieve a remission after Iomab-B or those who did not proceed to transplant, while the initial vertical drop in the curve in the control arm mainly represents patients who did not achieve a remission with salvage therapy and either crossed over to Iomab-B or went onto best supportive care.
Event-Free Survival with Iomab-B Versus Control Arm
The table below shows relevant adverse events in transplanted Iomab-B patients. In these patients, incidence of sepsis was four times lower in the Iomab-B arm than the control arm (6.1% vs. 28.6%). In addition, rates of other treatment related adverse events were lower in favor of Iomab-B, including febrile neutropenia (43.9% vs. 50.0%), mucositis (15.2% vs. 21.4%) and acute graft versus host disease (“GVHD”) (26.1% vs. 35.7%).
11
Grade ≥3 Treatment-Emergent Adverse Events in Transplanted Patients Through Day 100 Post-HCT

With current treatment practice, patients who have r/r AML with active disease, utilizing current conditioning agents have poor outcomes and very low survival rates. Using an Iomab-B led regimen, an unprecedented number of patients were able to access transplant and were able to do so with active disease, eliminating need for achieving a CR in order to transplant the patient. Thus, patients are also able to access BMT faster with Iomab-B, in less than half the time compared to conventional care. Iomab-B represents an exciting new paradigm with the potential to establish a new standard of care in r/r AML setting, making it possible for most patients to get to a successful transplant with Iomab-B with a portion of these patients having a long-term survival benefit. As shown below, with an Iomab-B led regimen, the majority of patients who are non-transplantable in routine clinical practice can be successfully transplanted, administering myeloablative radiation with reduced intensity conditioning tolerability to ultimately achieve transformative survival outcome, changing the treatment paradigm for r/r AML patients.
Iomab-B – New Paradigm to Upend BMT Access and Improve r/r AML Outcomes

Future Development and Life Cycle Management for Iomab-B
The results of the Pivotal Phase 3 SIERRA trial validate the value proposition of Iomab-B, and we believe it could establish unprecedented access to transplant (currently the only curative option) with better safety and tolerability and improved outcomes, all of which could potentially make Iomab-B the new standard of care for patients with r/r AML. We are actively working to launch an EAP and successfully file a BLA in the second half of 2023, and if approved, we anticipate the commercial launch for Iomab-B in 2024.
12
We intend to commercialize Iomab-B in the U.S. The commercial opportunity is supported by favorable dynamics, summarized by the “Three Ps and Two Cs”:
| ● | Patients: With its promising profile, Iomab-B provides the opportunity for unprecedented BMT access and better outcomes for patients, with favorable safety and tolerability |
| ● | Physicians: Our goal is to help physicians make BMT an option for a vast majority of r/r AML patients who currently are unable to access transplant without disruption to current practice. Patients are able to return to their referring physicians for post-BMT follow-up, long-term care |
| ● | Payers: Iomab-B potentially unlocks value through getting patients safely to effective, potentially curative transplants, with improved outcomes and a manageable safety and tolerability profile |
| ● | Competition: While there have been multiple new product approvals in AML over the last several years, they primarily focus on addressing genetic mutations, with limited competition in conditioning to increase access to BMT. We do not see direct or indirect visible competition for Iomab-B in the 5-to-10-year horizon to impair the commercial success of Iomab-B |
| ● | Concentrated Call Points: The commercialization for Iomab-B will benefit from a concentrated market. The top 50 centers perform 75% of BMTs and tend to be concentrated in metropolitan areas. These factors allow for commercialization delivered by a focused 35–50-person commercial organization. |
The favorable commercial dynamics for Iomab-B in the U.S. are further supported by the strong foundation of core competencies developed during the successful execution of the SIERRA trial at leading high-volume BMT centers. We established and actively managed end-to-end supply chain, never missing a patient dose, and were able to treat 60% more patients than expected due to the high number of crossover patients. We focused on operational excellence at the point of care, working in partnership with leading Key Opinion Leaders and their teams to successfully execute SIERRA at a wide array of centers. As a result, we have broad reach across leading BMT centers that account for 30% of BMT volume, which speaks to the concentration of the BMT market. The positive SIERRA results of unprecedented access and outcomes along with our commitment to operational excellence provide a strong foundation for our commercial team.
In April 2022, Actinium licensed the EUMENA commercial rights for Iomab-B to Immedica, an independent pharmaceutical company headquartered in Sweden. Immedica has significant know-how and experience in commercializing niche and specialty care products across Europe and the Middle East, with extensive regulatory and commercial expertise and capabilities. Actinium will continue to be responsible for certain clinical development activities and Iomab-B manufacturing and will retain commercialization rights in the U.S. and rest of the world. Currently, there an estimated ~7,200 BMTs for AML in EUMENA, two times that of the U.S., performed in a concentrated of number of centers. The incidence rate of AML in Europe is 3.7 per 100,000, or ~27,500 new patients per year. Actinium received an upfront payment of $35 million USD with the potential for an additional $417 million USD in regulatory and sales milestones and mid-twenty percent royalties. Iomab-B has been granted Orphan Drug Designation by the EMA and has received positive Scientific Advice from EMA that the SIERRA trial can support a marketing authorization with filing expected in 2024.
13
Background on Iomab-B
Iomab-B is a first in class targeted radiotherapy consisting of apamistamab, an anti-CD45 mouse antibody conjugated to radioactive iodine 131 (“I-131”) designed to deliver targeted myeloablative radiation to malignant and hematopoietic cells prior to allogeneic BMT. CD45 is uniquely expressed on blood cancer, immune and bone marrow stem cells at high levels. Targeting CD45 enables delivery of high radiation doses directly to the bone marrow, with a median of 16 gray and as high as 44.6 gray in the SIERRA trial, while minimizing radiation exposure to vital organs such as lungs, heart and gastrointestinal tract, thereby producing myeloablative outcomes with an overall better safety profile and the tolerability of a reduced intensity regimen. I-131 is a beta- and gamma-emitting radioisotope that works on the cell surface and does not need to be internalized. Developed at the Fred Hutchinson Cancer Research Center (“FHCRC”), Iomab-B has been studied in multiple disease indications including leukemias, lymphomas, MDS, and MM. Over 300 patients received Iomab-B through prior studies, demonstrating the potential for unprecedented access to BMT, improved survival and tolerability, and we intend to leverage these data as we plan for label expansion of Iomab-B. Iomab-B has been granted Orphan Drug Designation from the U.S. Food and Drug Administration (“FDA”) and has patent protection into 2037.
Actimab-A – CD33 targeting radiotherapeutic – mutation agnostic mechanism of action has potential as combination backbone therapy in highly radiosensitive, mutation rich AML
Our Actimab-A (225Ac-lintuzumab satetraxetan) program is focused on developing combinations with other AML treatment regimens with mechanistic synergies to establish Actimab-A as a backbone therapy, leveraging the mutation-agnostic mechanism of action of Actimab-A. There is no known resistance mechanism to targeted radiotherapies, making Actimab-A an attractive candidate for a variety of combinations. The scientific rational is to use CLAG-M, a powerful chemotherapy regimen routinely used to treat patients with r/r AML, and then use Actimab-A for its precision targeting ability that produces double-strand-DNA breaks that lead to cancer cell death to clear out residual disease. Actimab-A has demonstrated clinically significant survival benefit in a proof-of-concept study and is poised for advanced development in collaboration with the NCI.
Actimab-A + CLAG-M Phase 1 Study Results
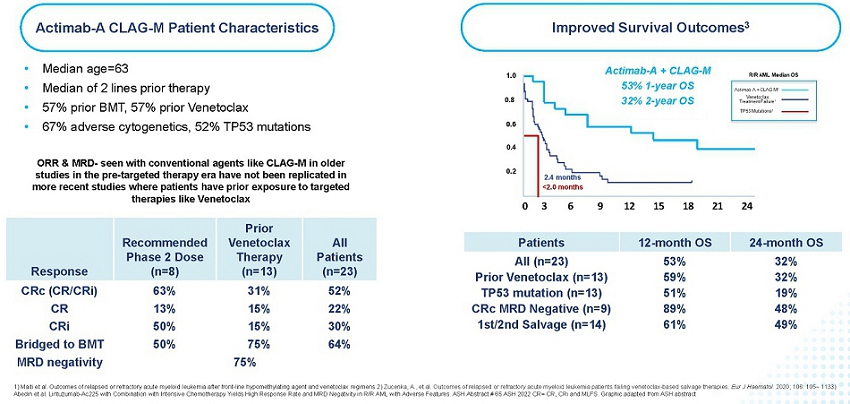
In collaboration with the Medical College of Wisconsin, the Actimab-A + CLAG-M Phase 1 trial was conducted in r/r AML patients. These patients had a median age of 63, failed two or more lines of therapy, which includes 57% having received prior treatment with venetoclax, a BCL-2 inhibitor. 67% of these patients had adverse cytogenetics, 52% had a TP53 mutation, and 57% had a prior BMT. Median OS is typically two to four months for this patient population, with a median OS of less than 3 months for patients who relapsed following venetoclax and a median OS less than 2 months for those with a TP53 mutation.
These trial results were presented as an oral presentation at the ASH Annual Meeting on December 10, 2022. In this difficult-to-treat r/r AML population, the results demonstrate its high potential. We reported 1-year survival of 53% and 2-year survival of 32%, which are as much as double what can be expected with currently available therapies. The trial showed an Overall Response Rate (“ORR”) of 65% across all dose cohorts, 52% complete remission rate, and a 75% MRD negativity rate. As highlighted in the figure below, the results are highly encouraging and show that the high rates of responses and MRD negativity are translating to a meaningful survival benefit in these difficult-to-treat patients, who would otherwise have dismal outcomes.
Actimab-A + CLAG-M – Impressive Response and Survival Benefit in r/r AML
14
Actimab-A + CLAG-M Compared to CLAG-M Alone in r/r AML
Efficacy of CLAG-M has been reported in older studies (Halpern and Walter. CLAG-M with dose-escalated mitoxantrone for adults with acute myeloid leukemia. Oncotarget 2018 and Mushtaq et al. Comparison of Salvage Chemotherapy Regimens in Relapsed/Refractory Acute Myeloid Leukemia. ASH 2018) in patients with r/r AML, however, almost all of these studies were conducted in the pre-targeted therapy era where no patients enrolled had prior venetoclax-based therapy, thus efficacy data of CLAG-M in the current era, in patients exposed to prior venetoclax, or with other high-risk features, is limited. When combined with Actimab-A, the combination has demonstrated a clinically significant survival benefit in a proof-of-concept study irrespective of prior targeted treatment. Relapsed or refractory AML after failing venetoclax-based therapy is associated with dismal survival outcomes, with a median OS of less than 3 months. In comparison, the combination trial of Actimab-A + CLAG-M led to 1-year survival of 59% and 2-year survival of 32% in patients who failed prior venetoclax-based therapy, which compares very favorably to the traditional outcomes in these patients.
Actimab-A + venetoclax Phase 1/2 Study Results
We are conducting a Phase 1/2 multi-center trial combining Actimab-A + venetoclax in both fit and unfit patients 18 years and older with r/r AML led by UCLA Medical Center. On December 10, 2022, data from our Actimab-A + venetoclax combination trial was presented at the ASH Annual Meeting. We have demonstrated preclinically that combinations of Actimab-A and venetoclax have mechanistic synergies. Overexpression of MCL-1, an anti-apoptotic protein, is associated with resistance to venetoclax in AML. Actimab-A kills tumors cells with DNA double-strand breaks and downregulates MCL-1, which can (re-)sensitize AML cells or reduce tumor resistance to venetoclax. The Actimab-A + venetoclax combination has been well-tolerated with responses, including a CR and a partial response in early dose escalation cohorts. In our ongoing clinical trial, we are exploring the optimal dose of Actimab-A, as well as the dosing regimen of the combination. We expect to present proof-of-concept of this study in the second half of 2023.
Advanced Development and Planning for Actimab-A
On February 6, 2023, we announced that we entered into a CRADA with the NCI, part of the NIH, to develop Actimab-A for the treatment of patients with AML and other hematologic malignancies. The NCI will serve as the regulatory sponsor for any clinical trials mutually approved by both parties to study Actimab-A, and the CRADA will provide extensive support for and accelerate the development of Actimab-A alone or in combination with chemotherapy, immunotherapy, targeted agents and other novel combinations. The CRADA studies will be overseen by the NCI in collaboration with Actinium’s clinical development team, where Actinium has the right to review and approval all protocols and has full right to all data. This broad collaboration may accelerate our Actimab-A development efforts with access to NCI’s vast network of over 2,000 clinical trial sites and its Myelomatch program. Later this year, we will provide updates on our progress as we move into late-stage development with Actimab-A + CLAG-M, as well as other developments with our venetoclax combination trial as part of our backbone development strategy.
Background on Actimab-A
Actimab-A is an anti-CD33 antibody linked to the potent alpha-emitting radioisotope Ac-225. Actimab-A targets CD33, which is expressed in virtually all malignant cells in patients with AML regardless of cytogenetics or mutations and enables potent alpha radiation to be directed against radiosensitive AML cells. These cells have no known resistance or repair mechanisms when hit with the alpha particles from the Ac-225 isotope payload that cause double stranded DNA breaks. We believe Actimab-A is the first radiotherapeutic for r/r AML and has the unique value proposition of broad applicability, a differentiated mechanism of action, and targeted precision that is well-tolerated with minimal toxicity. Our CD33 development program is driven by data obtained from approximately 150 AML patients in 6 trials and demonstrated single agent activity with high response rates, but was also associated with prolonged neutropenia. A combination strategy was considered appropriate given the changing treatment landscape of AML; hence, based on presumed mechanistic synergies, an investigator initiated trial of Actimab-A + CLAG-M and a company-sponsored Actimab-A + venetoclax were developed and patients were enrolled into these studies.
15
Conditioning Focused Programs
Iomab-B
We will further expand the Iomab-B franchise by focusing on lifecycle management for label enhancement and indication expansion. Iomab-B data in five additional hematologic indications (i.e., MDS, ALL, HL, NHL, and MM) provide the foundation to explore indication expansion opportunities to increase the total addressable market for Iomab-B. Across early trials at the FHCRC, Iomab-B demonstrated similar improved access to BMT and outcomes. We will leverage these data with strong results from the pivotal Phase 3 SIERRA trial to execute a comprehensive life cycle management strategy to further expand Iomab-B’s role in a variety of malignant and non-malignant hematological disorders, We will continue to develop the Iomab-B franchise to potentially address a broader market opportunity to address the over 165,000 patients diagnosed with cancers (e.g., leukemia, lymphoma, and myeloma), who could potentially benefit from transplant, but are unable to access one today.
Iomab-ACT
Iomab-ACT is comprised of apamistamab, the same anti-CD45 antibody as Iomab-B, but utilizes lower, nonmyeloablative levels of I-131 to achieve lymphodepletion for cellular therapies such as CAR-T or reduced intensity conditioning for gene therapies. We intend to continue to develop the Iomab-ACT program designed specifically for use prior to CAR-T and gene therapies, ultimately with a value proposition of improving overall access and outcomes for patients who need cellular or gene therapies.
Preclinical data showed a single, low-dose of Iomab-ACT demonstrated lymphodepletion and as CD45 positive immune cells are implicated in major CAR-T side effects, i.e., cytokine release syndrome ("CRS") and immune effector cell–associated neurotoxicity syndrome (“ICANS"), Iomab-ACT has the potential to be developed as a conditioning agent for CAR-T therapies. CRS and ICANS remain two most common toxicities of CAR-T therapies with severe cases (>Grade 3) seen in >20% of patients and fatality rates between 0-10%. Due to its effect on host monocytes/macrophages, we believe conditioning with Iomab-ACT will potentially reduce the incidence of CRS and ICANS.
Unlike chemotherapy, Iomab-ACT is targeted in nature, and we expect it to potentially promote improved CAR-T cell expansion, resulting in responses that are higher and more durable. We believe our Iomab-ACT program is highly differentiated when compared to fludarabine and cyclophosphamide (“Flu/Cy”) or other chemotherapy-based regimens that are used as standard practice today for lymphodepletion prior to cell therapy.
We are studying Iomab-ACT in collaboration with MSKCC, for conditioning prior to CAR-T therapy for patients with relapsed or refractory B-cell acute lymphoblastic leukemia (“B-ALL”) or diffuse large B-cell lymphoma (“DLBCL”). This study funded by a NIH grant is the first-of-its-kind study to use a radiotherapeutic-based conditioning regimen with CAR-T therapy. We have completed treatment of an initial cohort of three patients and will expand to a second cohort. This study was presented at the ASH Annual Meeting in December 2022 as a trial-in-progress. We expect to present proof-of-concept data from this study in 2023 and look forward to sharing more on our Iomab-ACT trial with MSKCC, along with future development plans in the CAR-T space.
R&D and Preclinical Programs
Our R&D efforts yield differentiated, high-value programs that demonstrate our experience across multiple validated cancer targets and isotopes and cover broad areas of focus leveraging our clinical development experience across hematology, targeted conditioning, solid tumors, and next generation radiotherapies. Our programs also inform the advancement of our Iomab-B, Actimab-A, and Iomab-ACT programs. Our research collaborations with Astellas, LG Chem, formerly AVEO Oncology, and EpicentRx establish our work with immunotherapies and in solid tumors. We are working on several preclinical programs which include novel approaches to established targets such as HER2 and HER3, as well as novel targets that show immense potential for radiotherapeutic approaches. Underpinning our development programs is our expanded patent portfolio of over 200 issued patents and pending patent applications worldwide.
16
We have utilized our technology platform to develop our clinical portfolio in hematology – Iomab-B and Actimab-A, in conditioning for transplant and as a therapeutic, respectively. In addition, our robust platform been used to develop a pipeline of novel radiotherapeutic assets to drive company growth. Preclinical pharmacology studies with our targeted radiotherapeutics such as HER3-ARC, HER2-ARC or CD33-ARC have shown dramatic improvement in tumor growth inhibition in various preclinical tumor models as single agents or in combination with immunotherapy such as magrolimab, an anti-CD47 monoclonal antibody. These results have prompted the team to spearhead efforts in multiple solid tumor programs.
Actinium’s lead solid tumor program is a targeted radiotherapy against HER3, a pan-cancer target that is overexpressed in several solid tumor indications with high unmet need. In April 2022, we presented the first HER3-targeted radiotherapeutic agent at the American Association for Cancer Research (“AACR”) Annual Meeting showing potent tumor cell cytotoxicity, enhanced antitumor effects and significantly improved survival with an Ac-225 radiolabeled HER3 antibody compared to a naked HER3 antibody in a preclinical non-small cell lung cancer (“NSCLC”) model. We have also demonstrated the direct impact of targeted radiotherapy in modulating immune signals such as calreticulin upregulation to enhance tumor cell killing. Further, we have leveraged the immunomodulatory effect(s) of targeted radiotherapy in combination with CD47 targeting agents such as magrolimab for sustained tumor growth inhibition in mouse models of AML and NSCLC. These results were presented at the Society for Immunotherapy of Cancer (“SITC”) Annual Meeting held in 2021.
Our R&D team continues to expand on capabilities and technologies across therapeutic modalities, linker technologies and in vivo cancer models, and build significant visibility through presentations at key conferences and publications in journals of high impact. Our R&D efforts are centered on the advancement of our key programs with a robust “fast-to-clinic” approach in niche indications and are backed by an extensive IP estate that comprises over 200 patents and patent applications including the methods of Ac-225 production.
Our Platform Technology
Our proprietary technology platform is built on the core competency to produce targeted radiotherapeutics, and coupled with our know-how and IP, establishes our company in the development of isotope-agnostic, multi-targeted products that may address the treatment of hard-to-treat diseases. In our clinical and preclinical programs, we have utilized multiple isotopes including Ac-225, I-131 and Lutetium-177 directed at multiple targets in oncology and hematology such as CD45, CD33, HER3, among others. Our targeted radiotherapies combine the cell-killing ability of radiation via a radioisotope payload with a targeting agent, such as a monoclonal antibody.
In addition to developing targeted radiotherapies, we also own patents related to the manufacturing of Ac-225 in a cyclotron. We have expertise in utilizing the alpha emitting isotope Ac-225 including clinical experience in treating approximately 150 patients with our alpha-emitter-based therapies, “gold standard” linker technology and five issued patents in the U.S. and 49 patents internationally related to the manufacturing or Ac-225 in a cyclotron, which we believe has the potential to produce higher quantities of highly pure Ac-225 than current methods. When appropriate, we are well-positioned to leverage this technology to produce Ac-225.
Intellectual Property
Our proprietary technology platform is supported by IP, know-how and trade secrets that cover the generation, development, methods of use and manufacture of targeted radiotherapies and their select components. Our IP covers various methods of use in multiple diseases, including indication, dose and scheduling, radionuclide warhead, and therapeutic combinations.
As of March 2023, we have expanded our patent portfolio to over 200 issued patents and pending patent applications worldwide, which we believe constitutes a valuable business asset. Our IP includes 45 patent families, including key patents that relate primarily to our radiotherapeutic candidates. Our patent portfolio includes 12 issued patents and 39 pending patent applications in the U.S., and 151 that are issued or pending internationally. The effective lives of the issued patents in our portfolio, or patents that may issue from the pending applications in our portfolio, ranges from expirations between 2024 and 2043.
For our Iomab-B product candidate, we have four issued patents in the U.S. and issued patents in Canada, Europe and Japan that relate to the composition. The basic patent terms of these patents expire in 2036 and 2037. Related patent applications are also currently pending in the U.S. and internationally. In addition, we own both U.S. and international pending patent applications that relate to the use of Iomab-B or Iomab-ACT in the treatment of cancers and non-malignant conditions.
17
Our patents also cover key areas of our business such as manufacturing key components of our product candidate, Actimab-A, including Ac-225 in a cyclotron. We have expertise in utilizing the alpha emitting isotope Ac-225 including clinical experience in treating approximately 150 patients with our alpha-emitter-based therapies, “gold standard” linker technology and five issued patents in the U.S. and 49 patents internationally related to the manufacturing or Ac-225 in a cyclotron, which we believe has the potential to produce higher quantities of Ac-225 than currently utilized methods. These patents will expire in the years 2024 through 2027. In addition, we also own U.S. and international patents and pending patent applications that relate to the manufacturing of Actimab-A and its use in the treatment of cancers.
Strategic Collaborations and Licensing Agreements
Fred Hutchinson Cancer Research Center
On June 15, 2012, the Company entered into a license and sponsored research agreement with FHCRC to build upon previous and ongoing clinical trials with Iomab-B. Developed at the FHCRC, a pioneer in the field of BMT, Iomab-B has been studied in over 400 patients and is supported by data in six disease indications including leukemias, lymphomas and multiple myeloma. The Company has been granted exclusive rights to the antibody and related master cell bank developed by FHCRC. A milestone payment of $1 million will be due to FHCRC upon FDA approval of the first drug utilizing the licensed antibody. Upon commercial sale of the drug, royalty payments of 2% of net sales will be due to FHCRC.
Immedica AB
In April 2022, Actinium licensed the EUMENA commercial rights for Iomab-B to Immedica, an independent pharmaceutical company headquartered in Sweden. Immedica has significant know-how and experience in commercializing niche and specialty care products across Europe and the Middle East, with extensive expertise and capabilities across sales and marketing, market access, regulatory and medical affairs, drug safety and quality assurance, among others. Actinium received an upfront payment of $35 million USD with the potential for an additional $417 million USD in regulatory and sales milestones and mid-twenty percent royalties. The market potential is extremely compelling in the EU with approximately 50% larger with 15,000 patients with r/r AML and double the number of BMTs performed than in the US. Iomab-B has been granted Orphan Drug Designation by the EMA and has received positive Scientific Advice from the Committee for Medicinal Products for Human Use (“CHMP”) of the EMA.
National Cancer Institute
In February 2023, we announced that Actinium entered into a CRADA with the NCI, part of the NIH, to develop Actimab-A in for the treatment of patients with AML and other hematologic malignancies. Under the terms of the CRADA, the NCI will serve as the regulatory sponsor for any clinical trials mutually approved by both parties to study Actimab-A while Actinium will be responsible for supplying and distributing Actimab-A to participating clinical sites and providing additional support as needed. The CRADA will provide broad support for the development of Actimab-A alone or in combination with chemotherapy, immunotherapy, targeted agents and other novel combinations. The CRADA studies will be overseen by NCI in collaboration with Actinium's clinical development team.
Astellas, LG Chem, and EpicentRx
We are leveraging our clinical experience, robust IP and radiotherapy know-how through research collaborations. Our collaborations with Astellas, LG Chem and EpicentRx establish our work with immunotherapies and in solid tumors. Through our research collaborations, such as with Astellas, we are advancing into solid tumors indications. With Astellas, we are utilizing a diagnostic agent developed in parallel with a therapeutic agent that shares the same target to identify patients who would benefit from the treatment.
Our platform is being utilized in our ongoing research collaboration with Astellas to arm select targeting agents owned by Astellas with the alpha-emitting radioisotope Ac-225 for the development of theranostics for solid tumor indications, which combine the ability of radioisotopes to be used for both diagnostic and therapeutic purposes.
18
We are also exploring novel targeted radiotherapies in solid tumors and blood cancers such as HER3 expressing solid tumors in collaboration with LG Chem and combinations with immunotherapies such as CD47 immune checkpoint inhibitors with EpicentRx.
Competition
The biopharmaceutical industry is extremely competitive and rapidly evolving, particularly in the field of oncology and hematology drug development. Our competition is likely to come from larger pharmaceutical companies, biotechnology companies, academia, and other public and private entities that focus on three broad areas relevant to our pipeline candidates – AML drugs, conditioning agents and radiopharmaceuticals. In addition, in markets where we are going after a target, companies with research programs and capabilities in our disease area focus may also be competing with our programs and pipeline.
In AML, the pipeline is crowded with 100+ programs, however, there are a few Phase 3 assets with limited potential that do not represent an imminent, competitive threat to Iomab-B or Actimab-A. None of the Phase 2 development programs in AML show the promise of producing high rates or duration of remission, and most patients that relapse tend to have poor survival outcomes. The Phase 2 assets primarily consist of agents targeting specific AML mutations, immunotherapies, or cell cycle modulators. Early clinical and preclinical stage assets consist of more cell therapy and immune cell engagers, and the potential success of these modalities in AML remain uncertain. Our strategy is to develop Actimab-A in combination with other products, and the agents in the development pipeline have the potential for synergies in combination with Actimab-A.
In conditioning, agents currently used for myeloablation prior to a BMT, lymphodepletion prior to CAR-T and other adoptive cell therapies and reduced intensity conditioning for gene therapy are largely generic, non-targeted chemotherapeutic agents. Recently, Jasper Therapeutics and Magenta Therapeutics ceased development of their antibody and antibody-drug conjugate or ADC conditioning programs for BMT. Certain companies such as Vertex Pharmaceuticals (“Vertex”), Gilead Sciences (“Gilead”) and Allogene Therapeutics (“Allogene") have or continue to explore non-chemotherapy conditioning with ADCs and antibodies for their in-house, proprietary cellular therapy programs. For example, Vertex has in-licensed ImmunoGen Inc.’s ADC technology, and had a collaboration with Molecular Templates, Inc. to develop targeted conditioning agents, which was subsequently terminated. Allogene is using its own proprietary anti-CD52 monoclonal antibody for use as a lymphodepletion agent in conjunction with CAR-T therapies. Telix Pharmaceuticals has announced plans for a conditioning program based on a CD66 radiotherapeutic approach in systemic amyloid light-chain amyloidosis (“SALA”) via an early stage investigator sponsored trial. Without exception, all these companies have either preclinical or early-stage programs that are solely focused on their proprietary programs. We believe that we are the only company with a phase 3 completed targeted conditioning asset that has demonstrated clinical benefit with the opportunity to be paradigm-changing.
Several companies are focused on developing radiotherapies, including, but not limited to: Abdera Therapeutics, Aktis Oncology, Bayer AG, Convergent Therapeutics, CuraSight, Fusion Pharmaceuticals, Inc., Lantheus Holdings, Inc., Mariana Oncology (previously, Curie Therapeutics), Monopar Therapeutics, Novartis AG, Point Biopharma, Inc., RadioMedix, Inc., Radiopharm Theranostics, Radionetics Oncology, RayzeBio, Inc., Q BioMed, Inc., Telix, and Y-mAbs Therapeutics, Inc.
Government Regulation
Regulatory Compliance
Our research and development activities are all subject to stringent regulation, primarily by the FDA in the U.S. under the Federal Food, Drug, and Cosmetic Act (the “FDCA”) and its implementing regulations, and the Public Health Service Act (“PHSA”) and its implementing regulations, and by comparable authorities under similar laws and regulations in other countries. This includes research and development, testing, and oversight of suppliers and contract manufacturers involved in the production of our product candidates we are developing, as well as the design, manufacturing, safety, efficacy, handling, labeling, storage, record-keeping, advertising, promotion and marketing. If, for any reason, we do not comply with applicable requirements, such noncompliance can result in adverse consequences, including delays in approval of, or even the refusal to approve, product licenses or other applications, the suspension or termination of clinical investigations, the revocation of approvals previously granted, as well as fines, criminal prosecution, recall or seizure of products, injunctions against shipping products and suspension of production and/or refusals of government contracts.
19
FDA Review Process and Product Approval
Our product candidates are regulated as biologics and must be approved by the FDA before they may be marketed in the U.S. This process generally involves the following:
| ● | completion of preclinical studies in accordance with the FDA’s current Good Laboratory Practices (“GLP”) requirements; |
| ● | submission to the FDA of an IND, which must become effective before human clinical trials may begin and must be updated annually; |
| ● | approval by an independent Institutional Review Board (“IRB”) ethics committee at each clinical site before the trial is initiated; |
| ● | performance of adequate and well-controlled clinical trials to establish the safety, purity and potency of the proposed biologic, and its safety and efficacy for each indication, in accordance with good clinical practice (“GCP”); |
| ● | submission to the FDA of a BLA for a new biologic, after completion of all pivotal clinical trials; |
| ● | a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities to assess compliance with applicable current Good Manufacturing Practice (“cGMP”) regulations; |
| ● | potential FDA audit of the clinical trial sites that generated the data in support of the BLA; and |
| ● | FDA review and approval of a BLA for a new biologic, prior to any commercial marketing or sale of the product in the U.S. |
Clinical trials generally are conducted in three sequential phases, although they may overlap or be combined.
| ● | Phase 1 studies are designed to evaluate the safety, dosage tolerance, metabolism and pharmacologic actions of the investigational product in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness |
| ● | Phase 2 studies are conducted to preliminarily or further evaluate the effectiveness of the investigational product for a particular indication(s) in patients with the disease or condition under study, to determine dosage tolerance and optimal dosage, and to identify possible adverse side effects and safety risks associated with the product |
| ● | Phase 3 clinical trials generally involve a large number of patients at multiple sites designed to provide the data required to demonstrate the effectiveness of the product for its intended use, safety and to establish the benefit-risk relationship of the product and provide an adequate basis for product labeling |
The results of the preclinical and clinical testing, along with information regarding the manufacturing of the product and proposed product labeling, are evaluated and, if determined appropriate, submitted to the FDA through a BLA. Once the BLA submission has been accepted for filing, the FDA’s standard goal is to review applications within ten months of the filing date or, if the application relates to a drug that treats a serious condition and would provide a significant improvement in safety or effectiveness qualifying for Priority Review, six months from the filing date. The review process is often significantly extended by FDA requests for additional information or clarification.
The FDA offers certain programs, such as Breakthrough Designation (“BTD”) and Fast Track designation, designed to expedite the development and review of applications for products intended for the treatment of a serious or life-threatening disease or condition. For BTD, preliminary clinical evidence of the product indicates that it may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. The FDA may initiate review of sections of a BLA before the application is complete, and the product may be eligible for accelerated approval. However, receipt of BTD or Fast Track designation does not ensure that a product will be developed or approved on an expedited basis, or at all.
The FDA reviews the BLA to determine, among other things, whether the proposed product is safe, pure and potent, which includes determining whether it is effective for its intended use, and whether the product is being manufactured in accordance with cGMP, to assure and preserve the product’s identity, strength, quality, potency and purity. The FDA may refer an application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved, and applications for new molecular entities and original BLAs are generally discussed at advisory committee meetings unless the FDA determines that this type of consultation is not needed under the circumstances.
20
After the FDA evaluates the BLA and conducts inspections of manufacturing facilities, it may issue an approval letter or a complete response letter (“CRL”). An approval letter authorizes commercial marketing of the biologic with specific prescribing information for specific indications. A CRL indicates that the review cycle of the application is complete, but the FDA cannot grant approval. A CRL may require additional inspections, and/or other significant, expensive and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. The FDA could approve the BLA with a Risk Evaluation and Mitigation Strategy (“REMS”) to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling, development of adequate controls and specifications, or a commitment to conduct one or more post-market studies or clinical trials. Such post-market testing may include Phase 4 clinical trials and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
Post-Approval Requirements
Any products manufactured or distributed by us or on our behalf pursuant to FDA approvals are subject to continuing regulation by the FDA and certain state agencies, including requirements for record-keeping, reporting of adverse experiences with the biologic, submitting biological product deviation reports to notify the FDA of unanticipated changes in distributed products, establishment registration, compliance with cGMP standards, and certain state licensing requirements.
Additionally, any significant change in the approved product or in how it is manufactured, including changes in formulation or the site of manufacture, generally require prior FDA approval. The packaging and labeling of all products developed by us are also subject to FDA approval and ongoing regulation. Noncompliance with any regulatory requirements can result in, among other things, issuance of warning letters, civil and criminal penalties, seizures, and injunctive action. Accordingly, manufacturers must continue to maintain compliance with cGMP and other aspects of regulatory compliance. The commercial distribution of prescription drugs is subject to the Drug Supply Chain Security Act (“DSCSA”), which regulates the distribution of the products at the federal level and sets certain standards for federal or state registration and compliance of entities in the supply chain.
The DSCSA preempts certain previously enacted state laws and the pedigree requirements of the Prescription Drug Marketing Act (“PDMA”). Trading partners within the drug supply chain must now ensure certain product tracing requirements are met, and are required to exchange transaction information, transaction history, and transaction statements. Product identifier information (an aspect of the product tracing scheme) is also now required. The DSCSA requirements, development of standards, and the system for product tracing have been and will continue to be phased in over a period of years through 2023. In addition to new legislation, FDA regulations, guidance documents, and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our product candidates.
Orphan Drug Act
We have received Orphan Drug designation for Iomab-B and Actimab-A for patients with AML. Under the Orphan Drug Act, FDA may grant Orphan Drug designation to drugs intended to treat a rare disease or condition, which is generally defined as a disease or condition that affects fewer than 200,000 individuals in the U.S. Orphan Drug designation must be requested before submitting a BLA. In the U.S., Orphan Drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages, and user-fee waivers. Orphan Drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first BLA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA Orphan Drug designation is entitled to a seven-year exclusive marketing period in the U.S. for that product, for that indication. During the seven-year exclusivity period, FDA may not approve any other applications to market the same drug for the same orphan indication, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity or where the manufacturer of the approved product cannot assure sufficient quantities. As a result, there can be no assurance that our competitors will not receive approval of drugs or biologics that have a different active ingredient for treatment of the diseases for which our products and product candidates are targeted.
21
Pediatric Information
Under the Pediatric Research Equity Act (“PREA”), certain BLAs must contain data to assess the safety and efficacy of the drug or biologic for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The Food and Drug Administration Safety and Innovation Act (“FDASIA”), amended the FDCA to require that a sponsor who is planning to submit a marketing application for a drug that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan (“PSP”) within 60 days of an end of Phase 2 meeting or, if there is no such meeting, as early as practicable before the initiation of the Phase 3 or Phase 2/3 study. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver. The FDA may grant deferrals for submission of pediatric data or full or partial waivers. A sponsor can submit amendments to an initial PSP if changes to the pediatric plan need to be considered based on preclinical data collected, early phase clinical trials as well as other clinical development programs.
Foreign Regulation
In addition to regulations in the U.S., we are subject to foreign regulations governing clinical trials and commercial sales and distribution of our product candidates, and products being marketed outside of the U.S. We must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of our products in those countries. The approval process varies from country to country, and the time may be longer or shorter than required by the FDA for BLA licensure. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. As in the U.S., we are subject to post-approval regulatory requirements.
Other Regulatory Considerations
We are also subject to regulation under the Occupational Safety and Health Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, The Clean Air Act, and other current and potential future federal, state, or local regulations. Our research and development activities involve the controlled use of hazardous materials, chemicals, biological materials and various radioactive compounds. We believe that our procedures comply with the standards prescribed by state and federal regulations; however, the risk of injury or accidental contamination cannot be completely eliminated. We may also be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments where we may market our products and product candidates, if approved. These laws and regulations include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, data privacy and security, aggregate spend reporting, and product price advertising.
The federal Anti-Kickback Statute, which prohibits, among other things, persons and entities including pharmaceutical manufacturers from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, overtly or covertly, in case or in kind, to induce or reward, or in return for, or either the referral of an individual for, or the purchase, lease or order or recommendation of an item or service reimbursable, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. The failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances.
In addition, Patient Protection and Affordable Care Act of 2010, as amended (“ACA”) codified cash law that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act (“FCA”). The FCA prohibits individuals or entities from, among other things, knowingly presenting or causing the presentation of a claims payment to, or approval by, the federal government that are false, fictitious or fraudulent, or knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim to avoid, decrease or conceal an obligation to pay money to the federal government. Our activities relating to the reporting of wholesaler or estimated retail prices for our products, the reporting of prices used to calculate Medicaid rebate information and other information affecting federal, state and third-party reimbursement for our products, and the sale and marketing of our products, are subject to scrutiny under the FCA. State statutes and regulations equivalent or substantially similar to the federal laws may extend to items and services reimbursed by commercial insurers and/or by patients directly. State law equivalents to the Anti-Kickback Statute and False Claims Act may not have adopted exceptions and safe harbors available at the federal level and therefore, may implicate a broader range of activities.
22
The federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) imposes criminal and civil liability for knowingly and willfully executing, or attempting to execute, a scheme to defraud or obtain, by any means of false or fraudulent pretenses, representations or promises, any money or property owned by, or under the control or custody of, any healthcare benefit program, including private third-party payors, and knowingly and willfully falsifying, concealing or covering up by trick, scheme or device, a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. The federal physician payment transparency requirements, sometimes referred to as the “Physician Payments Sunshine Act,” created under the ACA, and its implementing regulations, which requires applicable manufacturers of covered drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the State Children’s Health Insurance Program (with certain exceptions) to annually report to the Department of Health and Human Services (“HHS”), information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Under recent legislation, the Sunshine Act will extend to payments and transfers of value to physician assistants, nurse practitioners, and other mid-level healthcare providers. The Centers for Medicare and Medicaid Services (“CMS”) has the potential to impose penalties for violations of the Sunshine Act, depending on the circumstances, and payments reported under the Sunshine Act also have the potential to draw scrutiny on payments to and relationships with physicians and teaching hospitals, which may have implications under the Anti-Kickback Statute and other healthcare laws.
We may also be subject to data privacy and security regulation by both the federal government and the state governments in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act of 2009 (“HITECH”) and their respective implementing regulations, imposes, among other things, obligations, including mandatory contractual terms with respect to safeguarding the privacy, security and transmission of individually identifiable health information held by certain healthcare providers, health plans and healthcare clearinghouses, known as covered entities, and business associates. The HHS Office of Civil Rights (“OCR”) has increased its focus on compliance and continues to train state attorneys general for enforcement purposes. Even where HIPAA does not apply, according to the U.S. Federal Trade Commission (“FTC”), failing to take appropriate steps to keep consumers’ personal information secure constitutes unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act (“FTCA”), 15 U.S. Code §45(a). Medical data is considered sensitive data that merits stronger safeguards. There are numerous other laws and legislative and regulatory initiatives at the federal and state levels addressing privacy and security concerns, and some state privacy laws apply in broader circumstances than HIPAA.
We are subject to the U.S. Foreign Corrupt Practices Act (“FCPA”), which prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. Our present and future business has been and will continue to be subject to various other laws and regulations.
Human Capital
As of March 31, 2023, we had 49 full-time employees, 17 of whom have Ph.D. or M.D. degrees and 22 of whom are engaged in research and development and clinical development activities. We believe that we have been successful to date in attracting skilled and experienced personnel despite the competitive hiring marketing in the industry. Our employees are not covered by a collective bargaining agreement, and we believe that our relationship with our employees is excellent. We continue to engage external consultants on an as-needed basis to temporarily supplement existing staff.
Corporate Information
We were incorporated under the laws of the State of Delaware in 2013. Our principal executive offices are located at 275 Madison Avenue, New York, NY 10016, and our telephone number is (646) 677-3870. Our website address is www.actiniumpharma.com. The information contained on our website or that can be accessed through our website is not considered part of this report.
We make available free of charge through our website our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and any such amendments to those reports as soon as reasonably practicable after we electronically file such material with or furnish such material to the Securities and Exchange Commission (“SEC”). The SEC maintains a website at http://www.sec.gov that contains reports, proxy and information statements and other information regarding companies that file electronically with the SEC.
23
ITEM 1A. RISK FACTORS
In analyzing our company, you should consider carefully the following risk factors, together with all of the other information included in this Annual Report on Form 10-K. Factors that could cause or contribute to differences in our actual results include those discussed in the following subsection, as well as those discussed below in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere throughout this Annual Report on Form 10-K. The following are material factors that make an investment in our company speculative or risky. The risks and uncertainties described below are not the only ones we face. Additional risks not currently known to us or other factors not perceived by us to present significant risks to our business at this time also may impair our business operations.
Summary of Risk Factors
We are providing the following summary of the risk factors contained in this Annual Report on Form 10-K to enhance the readability and accessibility of our risk factor disclosures. We encourage you to carefully review the full risk factors contained in this Annual Report on Form 10-K in their entirety for additional information regarding the material factors that make an investment in our securities speculative or risky. These risks and uncertainties include, but are not limited to, the following:
| ● | We are a clinical-stage company and have generated no revenue from commercial sales to date; | |
| ● | We have incurred net losses in every year since our inception and anticipate that we will continue to incur net losses in the future; | |
| ● | If we fail to obtain additional financing, we will be unable to continue or complete our product development or product commercialization and you will likely lose your entire investment; | |
| ● | We are highly dependent on the success of Iomab-B and the SIERRA trial and we may not be able to complete the necessary clinical development or our development efforts may not result in the data necessary to receive regulatory approval; | |
| ● | Our business could be adversely affected by the effects of health epidemics, including the global COVID-19 pandemic; | |
| ● | We have not demonstrated that any of our products are safe and effective for any indication and will continue to expend substantial time and resources on clinical development before any of our current or future product candidates will be eligible for FDA approval, if ever; | |
| ● | Our clinical trials may fail to demonstrate adequately the efficacy and safety of our product candidates, which would prevent or delay regulatory approval and commercialization; | |
| ● | Preliminary, Interim, and “top-line” data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.; | |
| ● |
Healthcare legislative reform measures intended to increase pressure to reduce prices of pharmaceutical products paid for by Medicare or, otherwise, affect the federal regulation of the U.S. healthcare system could have a material adverse effect our business, future revenue, if any, and results of operations; | |
| ● | We rely on third parties to conduct our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines or comply with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize our product candidates; | |
| ● | We currently depend on a single third-party manufacturer to produce our pre-clinical and clinical trial drug supplies. Any disruption in the operations of our current third-party manufacturer, or other third-party manufacturers we may engage in the future, could adversely affect our business and results of operations; |
24
| ● | Our product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential, or result in significant negative consequences; | |
| ● | Our patent position is highly uncertain and involves complex legal and factual questions. | |
| ● | The use of hazardous materials, including radioactive and biological materials, in our research and development efforts imposes certain compliance costs on us and may subject us to liability for claims arising from the use or misuse of these materials; | |
| ● | We are highly dependent on our key personnel, and the demand for talent in the biotechnology industry is highly competitive; if we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement or execute our business strategy; | |
| ● | Certain provisions of our Certificate of Incorporation and Bylaws and Delaware law make it more difficult for a third party to acquire us and make a takeover more difficult to complete, even if such a transaction were in our stockholders’ interest; and | |
| ● | Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited. |
Risks Related to Our Business
We are a clinical-stage company and have generated no revenue from commercial sales to date.
We are a clinical-stage biopharmaceutical company with a limited operating history. We have no products approved for commercial sale and have not generated any revenue from product sales to date. We will encounter risks and difficulties frequently experienced by early-stage companies in rapidly evolving fields. If we do not address these risks successfully, our business will suffer.
We have incurred net losses in every year since our inception and anticipate that we will continue to incur net losses in the future.
We are not profitable and have incurred losses in each period since our inception. As of December 31, 2022 and December 31, 2021, we had an accumulated deficit of $288.8 million and $255.7 million, respectively. We reported a net loss of $33.0 million and $24.8 million for the years ended December 31, 2022 and 2021, respectively. We expect to continue to operate at a net loss as we continue our research and development efforts, continue to conduct clinical trials and develop manufacturing, sales, marketing and distribution capabilities. There can be no assurance that the products under development by us will be approved for sale in the United States or elsewhere. Furthermore, there can be no assurance that if such products are approved, they will be successfully commercialized, which would have an adverse effect on our business prospects, financial condition and results of operation.
If we fail to obtain additional financing, we will be unable to continue or complete our product development and you will likely lose your entire investment.
As of the date of filing this report, we expect that our existing resources will be more than sufficient to fund our planned operations for more than 12 months following the date of this report.
Our business or operations may change in a manner that would consume available funds more rapidly than anticipated and substantial additional funding may be required to maintain operations, fund expansion, develop new or enhanced products, acquire complementary products, business or technologies or otherwise respond to competitive pressures and opportunities, such as a change in the regulatory environment or a change in preferred cancer treatment modalities. However, we may not be able to secure funding when we need it or on favorable terms or indeed on any terms. In addition, from time to time, we may not be able to secure enough capital in a timely enough manner which may cause the generation of a going-concern opinion from our auditors which can and may impair our stock market valuation and also our ability to finance on favorable terms or indeed on any terms.
25
To raise additional capital, we may in the future offer additional shares of our common stock or other securities convertible into or exchangeable for our common stock. We cannot assure you that we will be able to sell shares or other securities in any other offering at a price per share that is equal to or greater than the price per share paid by investors, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders.
If we cannot raise adequate funds to satisfy our capital requirements, we will have to delay, scale back or eliminate our research and development activities, clinical studies or future operations. We may also be required to obtain funds through arrangements with collaborators, which arrangements may require us to relinquish rights to certain technologies or products that we otherwise would not consider relinquishing, including rights to future product candidates or certain major geographic markets. We may further have to license our technology to others. This could result in sharing revenues which we might otherwise have retained for ourselves. Any of these actions may harm our business, financial condition and results of operations.
The amount of funding we will need depends on many factors, including the progress, timing and scope of our product development programs; the progress, timing and scope of our preclinical studies and clinical trials; the time and cost necessary to obtain regulatory approvals; the time and cost necessary to further develop manufacturing processes and arrange for contract manufacturing; our ability to enter into and maintain collaborative, licensing and other commercial relationships; and our partners’ commitment of time and resources to the development and commercialization of our products.
We have limited access to the capital markets and even if we can raise additional funding, we may be required to do so on unfavorable terms.
We have limited access to the capital markets to raise funds. The capital markets have been unpredictable in the recent past for radioisotope and other oncology companies and unprofitable companies such as ours. In addition, it is generally difficult for development-stage companies to raise capital under current market conditions. The amount of capital that a company such as ours is able to raise often depends on variables that are beyond our control. As a result, we may not be able to secure financing on terms attractive to us, or at all. If we are able to consummate a financing arrangement, the amount raised may not be sufficient to meet our future needs. If adequate funds are not available on acceptable terms, or at all, our business, including our technology licenses, results of operations, financial condition and our continued viability will be materially adversely affected.
We are highly dependent on the success of Iomab-B and the SIERRA trial and we may not be able to complete the necessary clinical development or our development efforts may not result in the data necessary to receive regulatory approval.
We have completed patient enrollment in the pivotal Phase 3 SIERRA trial (Study of Iomab-B in Elderly Relapsed or Refractory AML), a 153-patient multi-center randomized trial that will compare outcomes of patients who receive Iomab-B and a BMT to those patients receiving physician’s choice of salvage chemotherapy, defined as conventional care, as no standard of care exists for this patient population. We have announced that Iomab-B met the primary endpoint of dCR in the SIERRA trial with statistical significance (p<0.0001). The SIERRA trial may be unsuccessful and fail to demonstrate a safety and efficacy profile that is necessary to receive favorable regulatory approval. Even if Iomab-B receives favorable regulatory approval, we may not be successful in securing adequate reimbursement or establishing successful commercial operations. Any or all of these factors could have a material adverse impact on our business and ability to continue operations.
We may be unable to establish sales, marketing and commercial supply capabilities.
We do not currently have, nor have we ever had, commercial sales and marketing capabilities. If any of our product candidates become approved, we would have to build and establish these capabilities in order to commercialize our approved product candidates. The process of establishing commercial capabilities will be expensive and time consuming. Even if we are successful in building sales and marketing capabilities, we may not be successful in commercializing any of our product candidates. Any delays in commercialization or failure to successfully commercialize any product candidate may have material adverse impacts on our business and ability to continue operations.
26
Our business could be adversely affected by the effects of health epidemics, including the global COVID-19 pandemic.
The global health crisis caused by the novel coronavirus COVID-19 pandemic and its resurgences has and may continue to negatively impact global economic activity, which, despite vaccination efforts, remains uncertain and cannot be predicted with confidence. In addition, highly transmissible new variants of COVID-19 have spread globally. The full impact of such variants cannot be predicted at this time, and could depend on numerous factors, including vaccination rates among the population, the effectiveness of COVID-19 vaccines and boosters against the COVID-19 variants and the response by governmental bodies and regulators. Given the ongoing and dynamic nature of the circumstances, it is difficult to predict the impact of the COVID-19 pandemic on our business.
Many countries around the world have imposed quarantines and restrictions on travel and mass gatherings and could reinstitute such policies in response to future COVID-19 outbreaks. In such a scenario, our ability to continue to operate our business may also be limited. Such events may result in a period of business, supply and drug product manufacturing disruption, and in reduced operations, any of which could materially affect our business, financial condition and results of operations. In response to COVID-19, we implemented hybrid working for our office-based staff, while our research staff has been actively working in our laboratory throughout the pandemic and thus far have not experienced a significant disruption or delay in our operations as it relates to the clinical development, preclinical development of manufacturing of our drug candidates. Although we are adhering to health and safety protocols, an outbreak of COVID-19 at our facilities could nonetheless cause shutdowns of facilities and a reduction in our workforce, which could cause a disruption or delay in such operations. New outbreaks may further divert the attention and efforts of the medical community to coping with COVID-19, and may disrupt the marketplace in which we operate and may have a material adverse effect on our operations.
A continuation or worsening of the levels of market disruption and volatility seen in the recent past could have an adverse effect on our ability to access capital, which could in the future negatively affect our liquidity. In addition, a recession or market correction resulting from the spread of COVID-19 could materially affect our business and the value of our common stock.
We believe our earlier stage CD33 clinical trials will continue to recruit and enroll patients given the acute nature of relapsed or refractory AML. The continuation of the pandemic could adversely affect our planned clinical trial operations, including our ability to conduct the trials on the expected timelines and recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 if their geography is impacted by the pandemic. Further, the continuation and/or resurgence of the COVID-19 pandemic could result in delays in our clinical trials due to prioritization of hospital resources toward the pandemic, restrictions in travel, potential unwillingness of patients to enroll in trials at this time, or the inability of patients to comply with clinical trial protocols if quarantines or travel restrictions are reinstated that impede patient movement or interrupt healthcare services. In addition, we rely on independent clinical investigators, contract research organizations and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our preclinical studies and clinical trials, and the pandemic may affect their ability to devote sufficient time and resources to our programs or to travel to sites to perform work for us, which may result in delays or hinder our ability to collect data from our clinical trials.
Additionally, COVID-19 may result in delays in receiving approvals from local and foreign regulatory authorities, delays in necessary interactions with IRB’s or Institutional Review Boards, local and foreign regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees.
We continue to monitor the impacts of COVID-19 on the global economy and on our business operations. However, the ultimate impact from COVID-19 on our business operations and financial results during 2023 will depend on, among other things, the ultimate severity and scope of the pandemic, including the new variants of the virus, and whether governmental and private travel restrictions and public concerns about public gatherings are reinstated. We are not able to fully quantify the impact that these factors had on our financial results during 2022 and will have in 2023.
27
Our business is subject to cybersecurity risks.
Our operations are increasingly dependent on information technologies and services. Threats to information technology systems associated with cybersecurity risks and cyber incidents or attacks continue to grow, and include, among other things, storms and natural disasters, terrorist attacks, utility outages, theft, viruses, phishing, malware, design defects, human error, and complications encountered as existing systems are maintained, repaired, replaced, or upgraded. Risks associated with these threats include, among other things:
| ● | theft or misappropriation of funds; | |
| ● | loss, corruption, or misappropriation of intellectual property, or other proprietary, confidential or personally identifiable information (including supplier, clinical data or employee data); | |
| ● | disruption or impairment of our and our business operations and safety procedures; | |
| ● | damage to our reputation with our potential partners, patients and the market; | |
| ● | exposure to litigation; | |
| ● | increased costs to prevent, respond to or mitigate cybersecurity events. |
Although we utilize various procedures and controls to mitigate our exposure to such risk, cybersecurity attacks and other cyber events are evolving and unpredictable. Moreover, we have no control over the information technology systems of third parties conducting our clinical trials, our suppliers, and others with which our systems may connect and communicate. As a result, the occurrence of a cyber incident could go unnoticed for a period time.
We have cybersecurity insurance coverage in the event we become subject to various cybersecurity attacks, however, we cannot ensure that it will be sufficient to cover any particular losses we may experience as a result of such cyberattacks. Any cyber incident could have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Regulation
The FDA or comparable foreign regulatory authorities may disagree with our regulatory plans and we may fail to obtain regulatory approval of our product candidates.
Our products are subject to rigorous regulation by the FDA and numerous other federal, state and foreign governmental authorities. The process of seeking regulatory approval to market an antibody radiation-conjugate product is expensive and time-consuming, and, notwithstanding the effort and expense incurred, approval is never guaranteed. If we are not successful in obtaining timely approval of our products from the FDA, we may never be able to generate significant revenue and may be forced to cease operations. In particular, the FDA permits commercial distribution of a new antibody radiation-conjugate product only after a BLA for the product has received FDA approval. The BLA process is costly, lengthy and inherently uncertain. Any BLA filed by us will have to be supported by extensive data, including, but not limited to, technical, preclinical, clinical trial, chemistry, manufacturing and controls (“CMC”) and labeling data, to demonstrate to the FDA’s satisfaction the safety and efficacy of the product for its intended use. The lengthy approval process as well as the unpredictability of future clinical trial results may result in our failing to obtain regulatory approval to market our product candidates, which would significantly harm our business, results of operations and prospects. In addition, even if we were to obtain approval, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may not approve the price we intend to charge for our products, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates.
28
In June 2012, we acquired rights to apamistamab, a clinical stage anti-CD45 monoclonal antibody with safety and efficacy data in more than 300 patients in need of a BMT. Iomab-B is our product candidate that links I-131 to apamistamab that is being studied in the pivotal Phase 3 SIERRA trial. Product candidates utilizing apamistamab would require BLA approval before they can be marketed in the United States. We are also evaluating Iomab-ACT, which uses a lower dose I-131 for lymphodepletion prior to CAR-T or adoptive cell therapy. We are currently evaluating clinical trials that would use our construct for lymphodepletion. Our CD33 Alpha program studying Actimab-A (lintuzumab-Ac-225) product candidate is also being studied in several Phase 1 trials under our sponsorship and investigator-initiated trials in patients with r/r AML. Product candidates utilizing the lintuzumab antibody would require BLA approval before they can be marketed in the United States. We are in the early stages of evaluating other product candidates consisting of conjugates of Ac-225 with human or humanized antibodies for pre-clinical and clinical development in other types of cancer. The FDA may not approve these products for the indications that are necessary or desirable for successful commercialization. The FDA may fail to approve any BLA we submit for new product candidates or for new intended uses or indications for approved products or future product candidates. Failure to obtain FDA approval for our products in the proposed indications would have a material adverse effect on our business prospects, financial condition and results of operations.
The approval process in the United States and in other countries could result in unexpected and significant costs for us and consume management’s time and other resources. The FDA and other foreign regulatory agencies could ask us to supplement our submissions, collect non-clinical data, conduct additional clinical trials or engage in other time-consuming actions, or it could simply deny our applications. In addition, even if we obtain approval to market our products in the United States or in other countries, the approval could be revoked, or other restrictions imposed if post-market data demonstrates safety issues or lack of effectiveness. We cannot predict with certainty how, or when, the FDA or other regulatory authorities will act. If we are unable to obtain the necessary regulatory approvals, our financial condition and cash flow may be materially adversely affected, and our ability to grow domestically and internationally may be limited. Additionally, even if we obtain approval, regulatory authorities may approve any of our product candidates for fewer or more limited indications that we request. The Company’s products may not be approved for the specific indications that are most necessary or desirable for successful commercialization or profitability.
We have not demonstrated that any of our products are safe and effective for any indication and will continue to expend substantial time and resources on clinical development before any of our current or future product candidates will be eligible for FDA approval, if ever.
We expect that a substantial portion of our efforts and expenditures over the next few years will be devoted to development of our existing and contemplated biological product candidates. Accordingly, our business currently depends heavily on the successful development, FDA approval, and commercialization of such candidates, which may never receive FDA approval or be successfully commercialized even if FDA approval is received. The research, testing, manufacturing, labeling, approval, sale, marketing, and distribution of our biological product candidates are, and will remain, subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, as applicable. We are currently not permitted to market any of our current or future product candidates in the United States until we receive FDA approval (of each) via the BLA process. To date, we have two product candidates in clinical development and have not-yet submitted a BLA for any of our candidates and, for many such candidates, do not expect to be in a position to do so for the foreseeable future, as there are numerous developmental steps that must be completed before we can prepare and submit a BLA.
29
In the United States, the FDA regulates pharmaceutical and biological product candidates under the FDCA and the Public Health Service Act (“PHSA”), as well as their respective implementing regulations. Such products and product candidates are also subject to other federal, state, and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations requires the expenditure of substantial time and financial resources. The process required by the FDA before a drug or biological product may be marketed in the United States generally involves the following:
| ● | completion of preclinical laboratory tests and animal studies in accordance with FDA’s good laboratory practices (“GLPs”) and applicable requirements for the humane use of laboratory animals or other applicable regulations; | |
| ● | submission to the FDA of an Investigational New Drug (“IND”), which must become effective before human clinical trials in the United States may begin; | |
| ● | performance of adequate and well-controlled human clinical trials in accordance with FDA’s IND regulations, GCPs, and any additional requirements for the protection of human research subjects and their health information, to establish the safety and efficacy of the proposed biological product for its intended use; | |
| ● | submission to the FDA of a BLA for marketing approval that meets applicable requirements to ensure the continued safety, purity, and potency of the product that is the subject of the BLA based on results of preclinical testing and clinical trials; | |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biological product is produced, to assess compliance with cGMPs and assure that the facilities, methods and controls are adequate to preserve the biological product’s identity, strength, quality and purity; | |
| ● | potential FDA audit of the nonclinical study and clinical trial sites that generated the data in support of the BLA; and | |
| ● | FDA review and approval, or denial, of the BLA. |
Before testing any biological product candidate in humans, the product candidate enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of the preclinical tests must comply with federal regulations and requirements including GLPs. The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials and places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a biological product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin or that, for those that have already commenced under an active IND, that issues will not arise that suspend or terminate such trials.
30
Clinical trials involve the administration of the biological product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations composing the GCP requirements, including the requirement that all research subjects provide informed consent. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase 1. The biological product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in subjects. | |
| ● | Phase 2. The biological product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. | |
| ● | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk to benefit ratio of the product and provide an adequate basis for product labeling. |
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up.
After the completion of clinical trials of a biological product, FDA approval of a BLA must be obtained before commercial marketing of the biological product. The BLA must include results of product development, laboratory and animal studies, human trials, information on the manufacture and composition of the product, proposed labeling and other relevant information. The FDA may grant deferrals for submission of data, or full or partial waivers. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all. Before approving a BLA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND trial requirements and GCP requirements. To assure cGMP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. We cannot predict with any certainty if or when we might submit a BLA for regulatory approval for our product candidates or whether any such BLA will be approved by the FDA. Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. For example, the FDA may not agree with our proposed endpoints for any clinical trial we propose, which may delay the commencement of our clinical trials. The clinical trial process is also lengthy and requires substantial time and effort.
In December 2015, the FDA cleared our IND filing for Iomab-B and we have completed patient enrollment of a randomized, controlled, pivotal Phase 3 clinical trial under such IND to study Iomab-B in patients 55 years of age or older with relapsed or refractory AML. The Phase 3 SIERRA trial met its primary endpoint with high statistical significance with positive results for secondary endpoints and exploratory endpoints and it is expected to form the basis for a BLA for Iomab-B for use in preparing and conditioning AML patients for a BMT. Additionally, there are physician IND trials at the FHCRC that have been conducted or are currently ongoing at FHCRC with Iomab-B (for other target indications) and the apamistamab antibody (formerly known as BC8) we licensed. We have other clinical trials ongoing and others that we have planned but not-yet commenced, for our other drug candidate Actimab-A under our own sponsorship and investigator-initiated trials ongoing. Except for Iomab-B (for patients with AML), we expect that the clinical trials we need to conduct to be in a position to submit BLAs for our product candidates currently in-development will take, at least, several years to complete. Moreover, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. Also, the results of early preclinical and clinical testing may not be predictive of the results of subsequent clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier studies. And, preclinical and clinical data are often susceptible to multiple interpretations and analyses. Many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have, nonetheless, failed to obtain marketing approval of their products. Success in preclinical testing and early clinical trials does not ensure that later clinical trials, which involve many more subjects, and the results of later clinical trials may not replicate the results of prior clinical trials and preclinical testing. Any failure or substantial delay in our product development plans may have a material adverse effect on our business.
31
We may encounter substantial delays in our clinical trials or may not be able to conduct our trials on the timelines we expect.
We cannot predict whether we will encounter problems with any of our ongoing or planned clinical trials that will cause us or regulatory authorities to delay, suspend, or discontinue clinical trials or to delay the analysis of data from ongoing clinical trials. Any of the following could delay or disrupt the clinical development of our product candidates and potentially cause our product candidates to fail to receive regulatory approval:
| ● | conditions imposed on us by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; | |
| ● | delays in receiving, or the inability to obtain, required approvals from IRBs or other reviewing entities at clinical sites selected for participation in our clinical trials; | |
| ● | delays in enrolling patients into clinical trials; | |
| ● | a lower than anticipated retention rate of patients in clinical trials; | |
| ● | the need to repeat or discontinue clinical trials as a result of inconclusive or negative results or unforeseen complications in testing or because the results of later trials may not confirm positive results from earlier preclinical studies or clinical trials; | |
| ● | inadequate supply, delays in distribution, deficient quality of, or inability to purchase or manufacture drug product, comparator drugs or other materials necessary to conduct our clinical trials; | |
| ● | unfavorable FDA or other foreign regulatory inspection and review of a clinical trial site or records of any clinical or preclinical investigation; | |
| ● | serious and unexpected drug-related side effects experienced by participants in our clinical trials, which may occur even if they were not observed in earlier trials or only observed in a limited number of participants; | |
| ● | a finding that the trial participants are being exposed to unacceptable health risks; | |
| ● | the placement by the FDA or a foreign regulatory authority of a clinical hold on a trial; or | |
| ● | delays in obtaining regulatory agency authorization for the conduct of our clinical trials. |
We may suspend, or the FDA or other applicable regulatory authorities may require us to suspend, clinical trials of a product candidate at any time if we or they believe the patients participating in such clinical trials, or in independent third-party clinical trials for drugs based on similar technologies, are being exposed to unacceptable health risks including but not limited to unacceptable or suboptimal factors related to toxicity, clinical efficacy, imbalances in safety and efficacy profiles or for other reasons.
Further, individuals involved with our clinical trials may serve as consultants to us from time to time and receive stock options or cash compensation in connection with such services. If these relationships and any related compensation to the clinical investigator carrying out the study result in perceived or actual conflicts of interest, or the FDA concludes that the financial relationship may have affected interpretation of the study, the integrity of the data generated at the applicable clinical trial site may be questioned and the utility of the clinical trial itself may be jeopardized. The delay, suspension or discontinuation of any of our clinical trials, or a delay in the analysis of clinical data for our product candidates, for any of the foregoing reasons, could adversely affect our efforts to obtain regulatory approval for and to commercialize our product candidates, increase our operating expenses and have a material adverse effect on our financial results.
32
Clinical trials may also be delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated by us, the FDA, the IRBs at the sites where the IRBs are overseeing a trial, or a data safety monitoring board, or DSMB (Data Safety Monitoring Board)/DMC (Data Monitoring Committee), overseeing the clinical trial at issue, or other regulatory authorities due to a number of factors, including:
| ● | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; | |
| ● | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; | |
| ● | varying interpretation of data by the FDA or similar foreign regulatory authorities; | |
| ● | failure to achieve primary or secondary endpoints or other failure to demonstrate efficacy; | |
| ● | unforeseen safety issues; or | |
| ● | lack of adequate funding to continue the clinical trial. |
Modifications to our product candidates may require federal approvals.
The BLA application is the vehicle through which the company may formally propose that the FDA approve a new pharmaceutical for sale and marketing in the United States. Once a particular product candidate receives FDA approval, expanded uses or uses in new indications of our products may require additional human clinical trials and new regulatory approvals, including additional IND and BLA submissions and premarket approvals before we can begin clinical development, and/or prior to marketing and sales. If the FDA requires new approvals for a particular use or indication, we may be required to conduct additional clinical studies, which would require additional expenditures and harm our operating results. If the products are already being used for these new indications, we may also be subject to significant enforcement actions.
Conducting clinical trials and obtaining approvals is a time-consuming process, and delays in obtaining required future approvals could adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would have an adverse effect on our business prospects, financial condition and results of operation.
Clinical trials necessary to support approval of our product candidates are time-consuming and expensive.
Initiating and completing clinical trials necessary to support FDA approval of a BLA for Iomab-B, Actimab-A, and other product candidates, is a time-consuming and expensive process, and the outcome is inherently uncertain. Moreover, the results of early clinical trials are not necessarily predictive of future results, and any product candidate we advance into clinical trials may not have favorable results in later clinical trials. We worked with the FDA to develop the SIERRA clinical trial to test the safety and efficacy of Iomab-B in patients with relapsed or refractory AML who are age 55 and above prior to a BMT. This trial is designed to support a BLA filing for marketing approval by the FDA. In addition to clinical data, a BLA filing encompasses preclinical, CMC, labeling and other information. Even if the clinical data from the SIERRA trial is positive, there can be no assurances that the BLA filing we produce will meet all of the FDA’s requirements or that they will not request additional information or studies, which may delay the FDA’s review or we may not be able to produce. We have also worked with the FDA to develop a regulatory pathway for lintuzumab-Ac-225 in patients with high-risk MDS that consists of a dose-confirming Phase 1 trial that can be followed by a randomized, controlled pivotal trial that could support a BLA filing. To date, we have not initiated this clinical trial and we may never elect or be able to do so. There can be no assurance that the data generated during the trial, or any trial, will meet our chosen safety and effectiveness endpoints or otherwise produce results that will eventually support the filing or approval of a BLA. Even if the data from this trial are favorable, the data may not be predictive of the results of any future clinical trials.
33
Preliminary, Interim, and “top-line” data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary, interim, and top-line data from our clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change as more patient data become available or following a more comprehensive review of the data related to the particular study or trial. For example, in October 2022 we announced that Iomab-B met the primary endpoint of dCR in the SIERRA trial with statistical significance (p<0.0001). We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. Our clinical trials may be open label studies and certain of our clinical development and or operations staff may review interim or preliminary safety or efficacy data during routine data collection, cleaning and analysis from time to time. Interim or preliminary results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results once additional data have been received and fully evaluated. Preliminary, interim or top-line data also remain subject to audit and verification procedures that may result in the final data being materially different from the top-line, interim or preliminary data we previously published. As a result, top-line, interim and preliminary data should be viewed with caution until the final data are available.
From time to time, we may also disclose interim data from our preclinical studies and clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Adverse differences between interim data and final data could significantly harm our business prospects. Further, disclosure of interim data by us or by our competitors could result in volatility in the price of our common stock.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is material or otherwise appropriate information to include in our disclosure.
If the interim, top-line or preliminary data that we report differ from final results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
Our clinical trials may fail to demonstrate adequately the efficacy and safety of our product candidates, which would prevent or delay regulatory approval and commercialization.
Even if our clinical trials are completed as planned, we cannot be certain that their results will support our product candidate claims or that the FDA or foreign authorities will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for the proposed indicated uses. If FDA concludes that the clinical trials for Iomab-B, Actimab-A, or any other product candidate for which we might seek approval, have failed to demonstrate safety and effectiveness, we would not receive FDA approval to market that product candidate in the United States for the indications sought. In addition, such an outcome could cause us to abandon the product candidate and might delay development of others. Any delay or termination of our clinical trials will delay or preclude the filing of any submissions with the FDA and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of a product candidate’s profile.
34
The intellectual property related to antibodies we have licensed has expired or likely expired.
The key patents related to the humanized antibody, lintuzumab, which we use in our Actimab-A product candidate have expired. It is generally possible that others may be eventually able to use an antibody with the same sequence, and we will then need to rely on additional patent protection covering alpha particle drug products comprising Ac-225. Our final drug construct, Actimab A, consists of the lintuzumab antibody labeled with the isotope Ac-225. We currently own issued and pending patents relating to methods of manufacturing Actimab-A, methods of treatment using Actimab-A and production of the Ac-225 isotope. In addition, we possess trade secrets and know how related to the manufacturing and use of isotopes. Any competing product based on the lintuzumab antibody is likely to require several years of development before achieving our product candidate’s current status and may be subject to significant regulatory hurdles but such development by others is nevertheless a possibility that could negatively impact our business in the future. We own 4 issued U.S. patents, 1 issued Canadian patent, 1 issued European patent (validated as a national patent in several countries) and 1 issued Japanese patent that relate to the composition of our Iomab-B product candidate. Patent applications relating to Iomab-B are also pending in the U.S. and internationally. We have and may continue to file patents related to Iomab-B that can provide barriers to entry but there is no certainty that these patents will be granted or such granting thereof will adequately prevent others from seeking to replicate and use the apamistamab antibody or the construct. Our patent portfolio includes pending applications related to radioimmunoconjugate composition, formulation administration, and methods of use in treating solid or liquid cancers. This subject matter includes composition, administration, and methods of treatment for our product candidates Actimab-A and Iomab-B. Any competing product based on the antibody used in Iomab-B is likely to require several years of development before achieving our product candidate’s current status and may be subject to significant regulatory hurdles but such development by others is nevertheless a possibility that could negatively impact our business in the future.
Our CD33 program clinical trials are testing the same drug construct.
Our CD33 program is comprised of several clinical trials including investigator-initiated trials in AML that are studying the same drug construct consisting of lintuzumab-Ac-225. Negative results from any of these trials could negatively impact our ability to enroll or complete our other trials studying lintzumab-Ac-225. Additionally, negative outcomes including safety concerns, may result in the FDA discontinuing other trials utilizing lintuzumab-Ac-225.
We may be unable to obtain a sufficient supply of isotopes to support clinical development or at commercial scale.
Iodine-131 is a key component of our Iomab-B drug candidate. We currently source medical grade I-131 from three suppliers including two leading global manufacturers. Currently, there is sufficient supply of I-131 to support additional trials we may undertake utilizing I-131 and for commercialization of Iomab-B. We continually evaluate I-131 manufacturers and suppliers and intend to have multiple qualified suppliers prior to the commercial launch of Iomab-B. While we consider I-131 to be commoditized and obtainable through several suppliers, there can be no guarantee that we will be able to secure I-131 or obtain I-131 on terms that are acceptable to us.
Actinium-225 is a key component of our Actimab-A product candidate, technology platform, preclinical R&D programs and other drug candidates that we might consider for development with the Ac-225 payload. There are adequate quantities of Ac-225 available today to meet our current needs via our present supplier, the Department of Energy (“DOE”). The Ac-225 currently supplied to Actinium’s clinical trials from the DOE is derived from the natural decay of thorium-229 from so-called ‘thorium-cows’ and is able to produce sufficient quantities that are several multiples of the amount of Ac-225 we require to supply our clinical programs through to early commercialization phase. The DOE is also producing Ac-225 from a recently developed alternative route for Ac-225 production via a linear accelerator that is currently being evaluated by Actinium. Initial preclinical and modelling results have indicated that the linear accelerator sourced Ac-225 does not impact labelling efficiency and expected distribution. In accordance with representations made by the DOE, the capacity of Ac-225 from this route is expected to be sufficient to supply all of Actinium’s pipeline and commercial Ac-225 needs and support new program expansion by not just Actinium but also other companies that are developing Ac-225 based products. Additional routes of Ac-225 production are being pursued by the DOE including the generation of new thorium cows and production via a cyclotron. The cyclotron production method for Ac-225 production leverages Actinium’s proprietary technology and know-how and presents an additional path towards production of high-quality Ac-225 at a scale that would be able to satisfy commercial needs. In addition, we are aware of at least ten other government and non-government entities globally including the U.S., Canada, Russia, Belgium, France and Japan that have, or expect to have ability to supply Ac-225 or equipment for its production within the timeframes relevant to the potential first commercial approval of our Ac-225-based drug candidate.
35
Our contract for supply of this isotope from the DOE must be renewed yearly, and we renewed our contract to extend through the end of 2023. While we expect this contract will continue to be renewed at the end of its term as it has since 2009, there can be no assurance that the DOE will renew the contract or that change its policies that allow for the sale of isotope to us. Failure to acquire sufficient quantities of medical grade Ac-225 would make it impossible to effectively complete clinical trials and to commercialize any Ac-225 based drug candidates that we may develop and would materially harm our business.
Our ability to conduct clinical trials to advance our drug candidates is dependent on our ability to obtain the radioisotopes I-131, Ac-225 and other isotopes we may choose to utilize in the future. Currently, we are dependent on third party manufacturers and suppliers for our isotopes. These suppliers may not perform their contracted services or may breach or terminate their agreements with us. Our suppliers are subject to regulations and standards that are overseen by regulatory and government agencies and we have no control over our suppliers’ compliance to these standards. Failure to comply with regulations and standards may result in their inability to supply isotopes and could result in delays in our clinical trials, which could have a negative impact on our business. We have developed intellectual property, know-how and trade secrets related to the manufacturing process of Ac-225. While we have manufactured medical grade Ac-225 of a purity compared to the cyclotron sourced material in the past, this activity was terminated due to operating cost reasons and we currently do not have experience in manufacturing medical grade Ac-225 and may not obtain the resources necessary to establish our own manufacturing capabilities in future. Our inability to build out and establish our own manufacturing facilities would require us to continue to rely on third party suppliers as we currently do. However, based on our current third-party suppliers and potential future suppliers of Ac-225 we expect to have adequate isotope supply to support our current ongoing clinical trials, current and planned preclinical R&D activities and commercialization should our drug candidates receive regulatory approval.
If we encounter difficulties enrolling patients in our clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
The timely completion of clinical trials in accordance with their protocols depends on our ability to enroll a sufficient number of patients who remain in the trial until its conclusion. We may experience difficulties in patient enrollment in our clinical trials for a variety of reasons, including:
| ● | the size and nature of the patient population; | |
| ● | the patient eligibility criteria defined in the protocol; | |
| ● | the size of the study population required for analysis of the trial’s primary endpoints; |
| ● | the proximity of patients to trial sites; | |
| ● | the design of the trial; | |
| ● | our ability to recruit clinical trial investigators with the appropriate competencies and expertise; | |
| ● | competing clinical trials for similar or alternate therapeutic treatments; | |
| ● | clinician’s and patients’ perceptions as to the potential advantages and side effects of the product candidate being studied in relation to other available therapies; | |
| ● | our ability to obtain and maintain patient consents; and | |
| ● | the risk that patients enrolled in clinical trials will not complete a clinical trial. |
In addition, refractory patients, which several of our trials are enrolling, participating in clinical trials are seriously and often terminally ill and therefore may not complete the clinical trial due to reasons including comorbid conditions or occurrence of adverse medical events related or unrelated to the investigational products, or death. Even if we are able to enroll a sufficient number of patients in our clinical trials, delays in patient enrollment will result in increased costs or affect the timing of our planned trials, which could adversely affect our ability to advance the development of our product candidates.
36
FDA may take actions that would prolong, delay, suspend, or terminate clinical trials of our product candidates, which may delay or prevent us from commercializing our product candidates on a timely basis.
There can be no assurance that the data generated in our clinical trials will be acceptable to FDA or that if future modifications during the trial are necessary, that any such modifications will be acceptable to FDA. Certain modifications to a clinical trial protocol made during the course of the clinical trial have to be submitted to the FDA. This could result in the delay or halt of a clinical trial while the modification is evaluated. In addition, depending on the quantity and nature of the changes made, FDA could take the position that some or all of the data generated by the clinical trial is not usable because the same protocol was not used throughout the trial. This might require the enrollment of additional subjects, which could result in the extension of the clinical trial and the FDA delaying approval of a product candidate. If the FDA believes that its prior approval is required for a particular modification, it can delay or halt a clinical trial while it evaluates additional information regarding the change.
Any delay or termination of our current or future clinical trials as a result of the risks summarized above, including delays in obtaining or maintaining required approvals from IRBs, delays in patient enrollment, the failure of patients to continue to participate in a clinical trial, and delays or termination of clinical trials as a result of protocol modifications or adverse events during the trials, may cause an increase in costs and delays in the filing of any submissions with the FDA, delay the approval and commercialization of our product candidates or result in the failure of the clinical trial, which could adversely affect our business, operating results and prospects. Lengthy delays in the completion of our Iomab-B clinical trials would adversely affect our business and prospects and could cause us to cease operations.
We have obtained orphan drug designation from FDA for two of our current product candidates and intend to pursue such designation for other candidates and indications in the future, but we may be unable to obtain such designations or to maintain the benefits associated with any orphan drug designations we have received or may receive in the future.
We have received orphan drug designation for Iomab-B and Actimab-A for treatment of AML in both the United States and the EU. Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the United States, or if it affects more than 200,000 individuals in the United States, there is no reasonable expectation that the cost of developing and making available a drug or biologic for this type of disease or condition will be recovered from sales in the United States for that drug or biologic. Similarly, the EMA grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention, or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the EU.
Orphan drug designation neither shortens the development time or regulatory review time of a drug or biologic nor gives the drug or biologic any advantage in the regulatory review or approval process. In the United States, orphan drug designation entitles a party to financial incentives, such as opportunities for grant funding towards clinical trial costs, tax advantages, and application fee waivers. In addition, if a product candidate receives the first FDA approval for the indication for which it has orphan designation, such product is entitled, upon approval, to seven years of orphan-drug exclusivity, during which the FDA may not approve any other application to market the same drug for the same indication, unless a subsequently approved product is clinically superior to orphan drug or where the manufacturer is unable to assure sufficient product quantity in the applicable patient population. In the EU, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity following drug or biological product approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Even if we obtain (or have obtained) orphan drug designation for certain product candidates, we may not be the first to obtain marketing approval for such candidates for the applicable indications due to the uncertainties inherent in the development of novel biologic products. And, an orphan drug candidate may not receive orphan-drug exclusivity upon approval if such candidate is approved for a use that is broader than the indication for which it received orphan designation. In addition, exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Finally, even if we successfully obtain orphan-drug exclusivity for an orphan drug candidate upon approval, such exclusivity may not effectively protect the product from competition because (i) different drugs with different active moieties can be approved for the same condition; and (ii) the FDA or EMA can also subsequently approve a subsequent product with the same active moiety and for the same indication as the orphan drug if the later-approved drug if deemed clinically superior to the orphan drug.
37
Even if we receive regulatory approval of our product candidates, we will be subject to ongoing regulatory obligations and continued regulatory review.
Any regulatory approvals that we receive for our product candidates will require surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require a REMS in order to approve our product candidates, which could entail requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. In addition, if the FDA or a comparable foreign regulatory authority approves our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for our product candidates will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMPs and GCPs for any clinical trials that we conduct post-approval. In addition, the FDA could require us to conduct another study to obtain additional safety or biomarker information. Later discovery of previously unknown problems with our product candidates, including adverse events of unanticipated severity or frequency, or with our third-party suppliers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| ● | restrictions on the marketing or manufacturing of our product candidates, withdrawal of the product from the market, or voluntary or mandatory product recalls; | |
| ● | fines, warning letters or holds on clinical trials; | |
| ● | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or suspension or revocation of license approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of our product candidates; and | |
| ● | injunctions or the imposition of civil or criminal penalties. |
The FDA’s and other regulatory authorities’ policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, and we may not achieve or sustain profitability.
Coverage and reimbursement may be limited or unavailable in certain market segments for our product candidates which could limit our sales of our product candidates, if approved.
The commercial success of our product candidates in both domestic and international markets will be substantially dependent on whether third-party coverage and reimbursement is available for patients that use our products. However, the availability of insurance coverage and reimbursement for newly approved cancer therapies is uncertain, and therefore, third-party coverage may be particularly difficult to obtain even if our products are approved by the FDA as safe and efficacious. Patients using existing approved therapies are generally reimbursed all or part of the product cost by Medicare or other third-party payors. Medicare, Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new drugs, and, as a result, they may not cover or provide adequate payment for these products. Submission of applications for reimbursement approval generally does not occur prior to the filing of a BLA for that product and may not be granted until many months after BLA approval. In order to obtain coverage and reimbursement for these products, we or our commercialization partners may have to agree to a net sales price lower than the net sales price we might charge in other sales channels. The continuing efforts of government and third-party payors to contain or reduce the costs of healthcare may limit our revenue. Initial dependence on the commercial success of our products may make our revenues particularly susceptible to any cost containment or reduction efforts.
38
Healthcare legislative reform measures intended to increase pressure to reduce prices of pharmaceutical products paid for by Medicare or, otherwise, affect the federal regulation of the U.S. healthcare system could have a material adverse effect our business, future revenue, if any, and results of operations.
In the United States, there have been a number of legislative and regulatory initiatives focused on containing the cost of healthcare. The Affordable Care Act, for example, substantially changed the way healthcare is financed by both governmental and private insurers. The Affordable Care Act contains a number of provisions that could impact our business and operations, primarily, once we obtain FDA approval to commercialize one of our product candidates in the United States, if ever, and may also affect our operations in ways we cannot currently predict. Affordable Care Act provisions that may affect our business include, among others, those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under health insurance exchanges, expansion of the 340B program, expansion of state Medicaid programs, fees and increased discount and rebate obligations, transparency and reporting requirements, and fraud and abuse enforcement. Such changes may impact existing government healthcare programs, industry competition, formulary composition, and may result in the development of new programs, including Medicare payment for performance initiatives, health technology assessments, and improvements to the physician quality reporting system and feedback program.
There have been significant judicial, administrative, executive, and legislative initiatives to modify, limit, replace, or repeal the Affordable Care Act since its enactment. For example, former President Trump issued several Executive Orders and other directives designed to delay the implementation of certain provisions of the Affordable Care Act or otherwise circumvent some of the requirements for health insurance mandated by the Affordable Care Act. Concurrently, Congress considered legislation that would repeal or replace all or part of the Affordable Care Act. While Congress has not passed comprehensive repeal legislation, several bills affecting the implementation the Affordable Care Act have been passed. For example, the Tax Cuts and Jobs Act of 2017 eliminated the Affordable Care Act provision requiring individuals to purchase and maintain health coverage, or the “individual mandate,” by reducing the associated penalty to zero, beginning in 2019. In December 2018, a district court in Texas held that the individual mandate is unconstitutional and that the rest of the Affordable Care Act is, therefore, invalid. On appeal, the Fifth Circuit Court of Appeals affirmed the holding on the individual mandate but remanded the case back to the lower court to reassess whether and how such holding affects the validity of the rest of the Affordable Care Act. The Fifth Circuit’s decision on the individual mandate was appealed to the U.S. Supreme Court. On June 17, 2021, the Supreme Court held that the plaintiffs (comprised of the state of Texas, as well as numerous other states and certain individuals) did not have standing to challenge the constitutionality of the Affordable Care Act’s individual mandate and, accordingly, vacated the Fifth Circuit’s decision and instructed the district court to dismiss the case. As a result, the Affordable Care Act will remain in-effect in its current form for the foreseeable future; however, we cannot predict what additional challenges may arise in the future, the outcome thereof, or the impact any such actions may have on our business.
In addition to the Affordable Care Act, there have been numerous other Congressional initiatives and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. Pharmaceutical product prices have been the focus of increased scrutiny by the government, including certain state attorneys general, members of Congress and the United States Department of Justice. State or federal healthcare reform measures or other social or political pressure to lower the cost of pharmaceutical products could have a material adverse impact on our business, results of operations and financial condition.
39
The Biden administration also introduced various measures in 2021 focusing on healthcare and drug pricing, in particular. For example, on January 28, 2021, President Biden issued an executive order that initiated a special enrollment period for purposes of obtaining health insurance coverage through the Affordable Care Act marketplace, which began on February 15, 2021, and remained open through August 15, 2021. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the Affordable Care Act. On the legislative front, the American Rescue Plan Act of 2021 was signed into law on March 11, 2021, which, in relevant part, eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source drugs and innovator multiple source drugs, beginning January 1, 2024. And, in July 2021, the Biden administration released an executive order entitled, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response, on September 9, 2021, HHS released a “Comprehensive Plan for Addressing High Drug Prices” that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles.
Most recently, on August 16, 2022, President Biden signed into law the Inflation Reduction Act of 2022 (the “IRA”), which, among other provisions, included several measures intended to lower the cost of prescription drugs and related healthcare reforms. Specifically, the IRA authorizes and directs the Department of Health and Human Services (the “DHHS”) to set drug price caps for certain high-cost Medicare Part B and Part D qualified drugs, with the initial list of drugs to be selected by September 1, 2023, and the first year of maximum price applicability to begin in 2026. The IRA further authorizes the DHHS to penalize pharmaceutical manufacturers that increase the price of certain Medicare Part B and Part D drugs faster than the rate of inflation. Finally, the IRA creates significant changes to the Medicare Part D benefit design by capping Part D beneficiaries’ annual out-of-pocket spending at $2,000 beginning in 2025. We cannot be sure whether additional or related legislation or rulemaking will be issued or enacted, or what impact, if any, such changes will have on the profitability of any of our drug candidates, if approved for commercial use, in the future.
Our relationships with customers, health care professionals and third-party payors may be subject to applicable healthcare laws, which could expose us to penalties, including administrative, civil or criminal penalties, damages, fines, imprisonment, exclusion from participation in federal healthcare programs such as Medicare and Medicaid, reputational harm, the curtailment or restructuring of our operations and diminished future profits and earnings.
Healthcare professionals and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our current and future arrangements with customers, healthcare professionals and third-party payors may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we conduct research, market, sell and distribute any products for which we obtain marketing approval. Federal and state healthcare laws and regulations that may affect our operations, directly or indirectly, include the following, among others:
| ● | the federal Anti-Kickback Statute, which prohibits persons and entities from, among other things, knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid; | |
| ● | the federal false claims laws, including civil whistleblower or qui tam actions under the federal False Claims Act, which impose criminal and civil penalties against individuals or entities for, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
40
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, which imposes criminal and civil liability for, among other things, executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and also imposes obligations, including mandatory contractual terms, on covered entities, including certain healthcare providers, health plans, and healthcare clearinghouses, and their respective business associates that create, receive, maintain or transmit individually identifiable health information for or on behalf of the covered entity as well as their covered subcontractors, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| ● | the federal Civil Monetary Penalties Law, which prohibits, among other things, the offering or transfer of remuneration to a Medicare or state healthcare program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state healthcare program, unless an exception applies; | |
| ● | the federal Physician Payments Sunshine Act, created under the Affordable Care Act, and its implementing regulations, which requires certain manufacturers of drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually information related to certain payments or other transfers of value provided to physicians and any ownership and investment interests held by physicians or their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding payments and other transfers of value to physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists and certified nurse midwives during the previous year; and | |
| ● | analogous state laws and regulations, including (among others) state anti-kickback and false claims laws, which may apply to our business practices, including, but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payor, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the United States federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information and that require tracking gifts and other remuneration and items of value provided to healthcare professionals and entities; state and local laws that require the registration of pharmaceutical sales representatives; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal law, thus complicating compliance efforts. |
Efforts to comply with applicable healthcare laws and regulations will involve substantial costs. Interpretations of standards of compliance under these laws and regulations are rapidly changing and subject to varying interpretations and it is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other laws that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, exclusion from government funded healthcare programs, such as Medicare and Medicaid, reputational harm, imprisonment, additional reporting obligations and oversight (if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws), and the curtailment or restructuring of our operations, any of which could diminish our future profits or earnings. If any of the physicians or other providers or entities with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
41
Third-party payors may not adequately reimburse customers for any product candidates that we may commercialize or promote, and may impose coverage restrictions or limitations such as prior authorizations and step edits that affect their use.
Our ability to commercialize any product candidates successfully also will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health programs, private health insurers, integrated delivery networks and other third-party payors. Third-party payors decide which medications they will pay for and establish reimbursement levels. A significant trend in the United States healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of payment for particular medications. Increasingly, third-party payors are requiring that drug companies provide predetermined discounts from list prices and are challenging the prices charged for medical products. Coverage and reimbursement may not be available for any product that we commercialize and, if reimbursement is available, the level of reimbursement may not be sufficient for commercial success. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If coverage and reimbursement is not available or is available only to limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval.
Obtaining reimbursement approval for any product candidate for which we obtain marketing approval from any government or other third-party payor is a time-consuming and costly process. There may be significant delays in obtaining coverage and adequate reimbursement for newly approved products. Moreover, eligibility for coverage and reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Even when a payor determines that a product that we may commercialize or promote is eligible for reimbursement under its criteria, the payor may impose coverage limitations that preclude payment for some uses that are approved by the FDA, or may impose restrictions, such as prior authorization requirements, or may simply deny coverage altogether. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Coverage and reimbursement rates may vary according to the use of the drug and the medical circumstances under which it is used may be based on reimbursement levels already set for lower cost products or procedures or may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Furthermore, the Centers for Medicare and Medicaid Services frequently change product descriptors, coverage policies, product and service codes, payment methodologies and reimbursement values. Commercial third-party payors often rely upon Medicare coverage policies and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain and maintain coverage and profitable payment rates from both government-funded programs and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize our approved products and our overall financial condition.
Risks Related to Third Parties
We rely on third parties to conduct our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines or comply with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize our product candidates.
We do not have the ability to independently conduct our clinical trials for our product candidates and we must rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories to conduct such trials. Our reliance on these third parties for clinical development activities results in reduced control over these activities. Moreover, the FDA requires us to comply with regulations and standards, commonly referred to as GCPs (good clinical practices), for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties does not relieve us of these responsibilities and requirements. If we or any of our third-party contractors fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials complies with GCP regulations. In addition, our clinical trials must be conducted with product produced under current good manufacturing practice, or cGMP, regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
42
If our consultants, contract research organizations and other similar entities with which we are working do not successfully carry out their contractual duties, meet expected deadlines, or comply with applicable regulations, we may be required to replace them. Although we believe that there are a number of other third-party contractors we could engage to continue these activities, we may not be able to enter into arrangements with alternative third-party contractors or to do so on commercially reasonable terms, which may result in a delay of our planned clinical trials and delayed development of our product candidates.
In addition, our third-party contractors are not our employees, and except for remedies available to us under our agreements with such third-party contractors, we cannot control whether or not they devote sufficient time and resources to our programs. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize, our product candidates on a timely basis, if at all, and our business, operating results and prospects would be adversely affected.
The antibodies we use in our targeted radiotherapy product candidates may be subject to generic competition.
We are not aware of any existing or pending regulations or legislation that pertains to generic radiopharmaceutical products such as our targeted radiotherapy product candidates. Our product candidates are regulated by the FDA as biologic products and we intend to seek approval for these products pursuant to the BLA pathway. The Biologics Price Competition and Innovation Act of 2009, or BPCIA, created an abbreviated pathway for the approval of biosimilar and interchangeable biologic products. The abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the original branded product was approved under a BLA. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. Even if a biosimilar gets approved for one of the antibodies that we use, the final constructs of our drug candidates consist of an antibody, radioisotope and in some cases a linker. Therefore, we do not believe that the final drug product of our candidates can be subject to competition from a biosimilar as outlined in BPCIA.
Our product candidates may never achieve market acceptance.
Iomab-B, Actimab-A product and future product candidates that we may develop may never gain market acceptance among physicians, patients and the medical community. The degree of market acceptance of any of our products will depend on a number of factors, including the actual and perceived effectiveness and reliability of the product; the results of any long-term clinical trials relating to use of the product; the availability, relative cost and perceived advantages and disadvantages of alternative technologies; the degree to which treatments using the product are approved for reimbursement by public and private insurers; the strength of our marketing and distribution infrastructure; and the level of education and awareness among physicians and hospitals concerning the product.
We believe that oncologists and other physicians will not widely adopt a product candidate unless they determine, based on experience, clinical data, and published peer-reviewed journal articles, that the use of that product candidate provides an effective alternative to other means of treating specific cancers. Patient studies or clinical experience may indicate that treatment with our product candidates does not provide patients with sufficient benefits in extension of life or quality of life. We believe that recommendations and support for the use of each product candidate from influential physicians will be essential for widespread market acceptance. Our product candidates are still in the development stage and it is premature to attempt to gain support from physicians at this time. We can provide no assurance that such support will ever be obtained. If our product candidates do not receive such support from these physicians and from long-term data, physicians may not use or continue to use, and hospitals may not purchase or continue to purchase, them.
Failure of Iomab-B, Actimab-A or any of our other product candidates to significantly penetrate current or new markets would negatively impact our business financial condition and results of operations.
43
We may be subject to claims that our third-party service providers, consultants or current or former employees have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial cost and be a distraction to our management and employees.
We currently depend on single third-party manufacturers to produce our pre-clinical and clinical trial drug supplies. Any disruption in the operations of our current third-party manufacturers, or other third-party manufacturers we may engage in the future, could adversely affect our business and results of operations.
We do not currently operate manufacturing facilities for pre-clinical or clinical production of any of our product candidates. We rely on third-party manufacturers to supply, store, and distribute pre-clinical and clinical supply of the components of our drug product candidates including monoclonal antibodies, linkers and radioisotopes, as well as the final construct which comprises our drug product candidates. We expect to continue to depend on third-party manufacturers for the foreseeable future. Any performance failure on the part of our existing or future manufacturers could delay clinical development, cause us to suspend or terminate development or delay or prohibit regulatory approval of our product candidates or commercialization of any approved products. Further avenues of disruption to our clinical or eventual commercial supply may also occur due to the sale, acquisition, business reprioritization, bankruptcy or other unforeseen circumstances that might occur at any of our suppliers or contract manufacturing partners including an inability to come to terms on renewal of existing contracts or new contracts.
We currently rely on single manufacturers to manufacture our pre-clinical and clinical trial drug supplies. With a view to maintaining business continuity we are evaluating alternatives and second and even third sources of supply or manufacturing for our core suppliers and manufacturing partners, however there can be no assurances that we will be able to identify such suppliers or partners and assuming we did, that we would be able to enter into contracts that are on favorable terms or on terms that will enable sufficient supply to ensure business continuity and support our growth plans.
Our product candidates require precise, high-quality manufacturing. Failure by our current contract manufacturer or other third-party manufacturers we may engage in the future to achieve and maintain high manufacturing standards could result in patient injury or death, product recalls or withdrawals, delays or failures in testing or delivery, cost overruns, or other problems that could seriously hurt our business. Contract manufacturers may encounter difficulties involving production yields, quality control, and quality assurance. These manufacturers are subject to ongoing periodic and unannounced inspections by the FDA and corresponding state and foreign agencies to ensure strict compliance with cGMPs and other applicable government regulations and corresponding foreign standards; we do not have control over third-party manufacturers’ compliance with these regulations and standards.
We may elect to build or purchase a manufacturing facility or facilities in the future to operate for the purposes of manufacturing our own products. We have never built, owned or operated a manufacturing facility. There can be no assurances that we will be able to successfully accomplish this and in doing so we may experience delays, cost overruns, or other problems that could seriously hurt our business. Even if we successfully build or purchase a manufacturing facility, we may not realize the expected benefits of these efforts.
We depend on vendors with specialized operations, equipment and know-how to manufacture the respective components of our drug candidates. We have entered into manufacturing and supply agreements with these third-parties, and in some instances, we have agreed that such vendor be the exclusive manufacturer and supplier. If any of the third-parties we depend on encounter difficulties in their operations, fail to comply with required regulations or breach their contractual obligations it may be difficult, or we may be unable to identify suitable alternative third-party manufacturers. While we identify and evaluate third-party manufacturers from time to time, even if we do identify suitable alternative third-parties, we may fail to reach agreement on contractual terms, it may be prohibitively expensive and there can be no assurance that we can successfully complete technology transfer and development work necessary or complete the necessary work in a timely manner. Any of which could prevent us from commencing manufacturing with third-parties which could cause delays or suspension of our clinical trials and pre-clinical work that may have a negative impact on our business.
44
Furthermore, these third-party contractors, whether foreign or domestic, may experience regulatory compliance difficulty, mechanical shutdowns, employee strikes, or any other unforeseeable acts that may delay or limit production. Our inability to adequately establish, supervise and conduct (either ourselves or through third parties) all aspects of the formulation and manufacturing processes, and the inability of third-party manufacturers to consistently supply quality product when required would have a material adverse effect on our ability to develop or commercialize our products. We have faced delays and risks associated with reliance on key third party manufacturers in the past and may be faced with such delays and risks in the future. Any future manufacturing interruptions or related supply issues could have an adverse effect on our company, including delays in clinical trials.
If we are successful in obtaining marketing approval from the FDA and/or other regulatory agencies for any of our product candidates, we anticipate continued reliance on third-party manufacturers.
To date, our product candidates have been manufactured in small quantities for preclinical and clinical testing by third-party manufacturers. If the FDA or other regulatory agencies approve any of our product candidates for commercial sale, we expect that we would continue to rely, at least initially, on third-party specialized manufacturers to produce commercial quantities of approved products. These manufacturers may not be able to successfully increase the manufacturing capacity for any approved product in a timely or economic manner, or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. Scale-up for commercial product may require financial commitment or investment by us, which we may not have sufficient capital for or may elect not to undertake. If third party manufacturers are unable to successfully increase the manufacturing capacity for a product candidate, or we are unable to establish our own manufacturing capabilities, the commercial launch of any approved products may be delayed or there may be a shortage in supply, which in turn could have a material adverse effect on our business.
In addition, the facilities used by our contract manufacturers to manufacture our product candidates must be approved by the FDA pursuant to inspections that will be conducted after we submit a BLA to the FDA. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with cGMPs. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or other regulatory authorities, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
We may have conflicts with our partners that could delay or prevent the development or commercialization of our product candidates.
We may have conflicts with our partners, such as conflicts concerning the interpretation of preclinical or clinical data, the achievement of milestones, the interpretation of contractual obligations, payments for services, development obligations or the ownership of intellectual property developed during our collaboration. If any conflicts arise with any of our partners, such partner may act in a manner that is adverse to our best interests. Any such disagreement could result in one or more of the following, each of which could delay or prevent the development or commercialization of our product candidates, and in turn prevent us from generating revenues: unwillingness on the part of a partner to pay us milestone payments or royalties we believe are due under a collaboration; uncertainty regarding ownership of intellectual property rights arising from our collaborative activities, which could prevent us from entering into additional collaborations; unwillingness by the partner to cooperate in the development or manufacture of the product, including providing us with product data or materials; unwillingness on the part of a partner to keep us informed regarding the progress of its development and commercialization activities or to permit public disclosure of the results of those activities; initiating litigation or alternative dispute resolution options by either party to resolve the dispute; or attempts by either party to terminate the agreement.
45
If in the future we are unable to establish U.S. or global sales and marketing capabilities or enter into agreements with third parties to sell and market our product candidates, we may not be successful in commercializing our product candidates if they are approved and we may not be able to generate any revenue.
We currently do not have a marketing or sales team for the marketing, sales and distribution of any of our product candidates that may receive regulatory approval. In order to commercialize any product candidates after approval, we must build on a territory-by-territory basis marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. If our product candidates receive regulatory approval, we may decide to establish an internal sales or marketing team with technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be expensive and time-consuming and will require significant attention of our executive officers to manage. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of any of our product candidates that we obtain approval to market.
With respect to the commercialization of all or certain of our product candidates, we may choose to collaborate, either globally or on a territory-by-territory basis, with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. In particular, we have and expect to continue to partner with third parties to commercialize Iomab-B outside the United States. In April 2022, we entered into a licensing agreement with Immedica, in which Immedica acquired the product rights for commercialization of Iomab-B for certain territories outside the U.S. If we are unable to enter into or maintain such arrangements when needed on acceptable terms, or at all, we may not be able to successfully commercialize any of our product candidates that receive regulatory approval or any such commercialization may experience delays or limitations. If we are not successful in commercializing our product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
We face significant competition from other biotechnology and pharmaceutical companies.
Our product candidates face, and will continue to face, intense competition from large pharmaceutical and biotechnology companies, as well as academic and research institutions. We compete in an industry that is characterized by (i) rapid technological change, (ii) evolving industry standards, (iii) emerging competition and (iv) new product introductions. Our competitors have existing products and technologies that will compete with our product candidates and technologies and may develop and commercialize additional products and technologies that will compete with our product candidates and technologies. Because several competing companies and institutions have greater financial resources than us, they may be able to (i) provide broader services and product lines, (ii) make greater investments in research and development, or R&D, and (iii) carry on broader R&D initiatives. Our competitors also have greater development capabilities than we do and have substantially greater experience in undertaking preclinical and clinical testing of product candidates, obtaining regulatory approvals, and manufacturing and marketing pharmaceutical products. They also have greater name recognition and better access to customers than us.
Our product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential, or result in significant negative consequences.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly. Even if any of our product candidates receives marketing approval, as greater numbers of patients use a product following its approval, an increase in the incidence of side effects or the incidence of other post-approval problems that were not seen or anticipated during pre-approval clinical trials could result in a number of potentially significant negative consequences, including:
| ● | regulatory authorities may withdraw their approval of the product; | |
| ● | regulatory authorities may require the addition of labeling statements, such as warnings or contraindications; | |
| ● | we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
46
| ● | we may elect, or we may be required, to recall or withdraw product from the market; | |
| ● | we could be sued and held liable for harm caused to patients; and | |
| ● | our reputation may suffer. |
Any of these events could substantially increase the costs and expenses of developing, commercializing and marketing any such product candidates or could harm or prevent sales of any approved products.
Risks Related to Our Intellectual Property
We depend upon securing and protecting critical intellectual property.
We are dependent on obtaining and maintaining patents, trade secrets, copyright and trademark protection of our technologies in the United States and other jurisdictions, as well as successfully enforcing this intellectual property and defending this intellectual property against third-party challenges. The degree of future protection of our proprietary rights is uncertain for product candidates that are currently in the early stages of development because we cannot predict which of these product candidates will ultimately reach the commercial market or whether the commercial versions of these product candidates will incorporate proprietary technologies.
Our patent position is highly uncertain and involves complex legal and factual questions.
Accordingly, we cannot predict the breadth of claims that may be allowed or enforced under our patents or in third-party patents. For example, we or our licensors might not have been the first to make the inventions covered by each of our pending patent applications and issued patents; we or our licensors might not have been the first to file patent applications for these inventions; others may independently develop similar or alternative technologies or duplicate any of our technologies; it is possible that none of our pending patent applications or the pending patent applications of our licensors will result in issued patents; our issued patents and issued patents of our licensors may not provide a basis for commercially viable technologies, or may not provide us with any competitive advantages, or may be challenged and invalidated by third parties; and, we may not develop additional proprietary technologies that are patentable.
As a result, our owned and licensed patents may not be valid, and we may not be able to obtain and enforce patents and to maintain trade secret protection for the full commercial extent of our technology. The extent to which we are unable to do so could materially harm our business.
We or our licensors have applied for and will continue to apply for patents for certain products and methods. Such applications may not result in the issuance of any patents, and any patents now held or that may be issued may not provide us with adequate protection from competition. Furthermore, it is possible that patents issued or licensed to us may be challenged successfully. In that event, if we have a preferred competitive position because of such patents, such preferred position would be lost. If we are unable to secure or to continue to maintain a preferred position, we could become subject to competition from the sale of generic products. Failure to receive, inability to protect, or expiration of our patents for medical use, manufacture, conjugation and labeling of Ac-225, the antibodies that we license from third parties, or subsequent related filings, would adversely affect our business and operations.
Patents issued or licensed to us may be infringed by the products or processes of others. The cost of enforcing our patent rights against infringers, if such enforcement is required, could be significant, and we do not currently have the financial resources to fund such litigation. Further, such litigation can go on for years and the time demands could interfere with our normal operations. There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical industry. We may become a party to patent litigation and other proceedings. The cost to us of any patent litigation, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation more effectively than we can because of their substantially greater financial resources. Litigation may also absorb significant management time.
47
Unpatented trade secrets, improvements, confidential know-how and continuing technological innovation are important to our scientific and commercial success. Although we attempt to and will continue to attempt to protect our proprietary information through reliance on trade secret laws and the use of confidentiality agreements with our partners, collaborators, employees and consultants and other appropriate means, these measures may not effectively prevent disclosure of our proprietary information, and, in any event, others may develop independently, or obtain access to, the same or similar information.
Certain of our patent rights are licensed to us by third parties. If we fail to comply with the terms of these license agreements, our rights to those patents may be terminated, and we may be unable to conduct our business.
If we are found to be infringing on patents or trade secrets owned by others, we may be forced to cease or alter our product development efforts, obtain a license to continue the development or sale of our products, and/or pay damages.
Our manufacturing processes and potential products may violate proprietary rights of patents that have been or may be granted to competitors, universities or others, or the trade secrets of those persons and entities. As the pharmaceutical industry expands and more patents are issued, the risk increases that our processes and potential products may give rise to claims that they infringe the patents or trade secrets of others. These other persons could bring legal actions against us claiming damages and seeking to enjoin clinical testing, manufacturing and marketing of the affected product or process. If any of these actions are successful, in addition to any potential liability for damages, we could be required to obtain a license in order to continue to conduct clinical tests, manufacture or market the affected product or use the affected process. Required licenses may not be available on acceptable terms, if at all, and the results of litigation are uncertain. If we become involved in litigation or other proceedings, it could consume a substantial portion of our financial resources and the efforts of our personnel.
In addition to infringement or other intellectual property claims against us, we may become a party to other patent litigation or proceedings before regulatory agencies, including post-grant review, inter parties review, interference or re-examination proceedings filed with the U.S. Patent and Trademark Office (or similar proceedings before corresponding tribunals in other jurisdictions) that challenge our patent rights or the patent rights of our licensors. The costs and efforts of defending our patents or enforcing our proprietary rights in post-issuance administrative proceedings can be substantial and the outcome can be uncertain. An adverse determination in these proceedings could weaken or invalidate the patent claims that cover our technology, which adverse determination could harm our business significantly and dissuade companies from collaborating with us or permit third parties to directly compete with the same technology.
Our ability to protect and enforce our patents does not guarantee that we will secure the right to commercialize our patents.
A patent is a limited monopoly right conferred upon an inventor, and his successors in title, in return for the making and disclosing of a new and non-obvious invention. This monopoly is of limited duration but, while in force, allows the patent holder to prevent others from making and/or using its invention. While a patent gives the holder this right to exclude others, it is not a license to commercialize the invention where other permissions may be required for commercialization to occur. For example, a drug cannot be marketed without the appropriate authorization from the FDA, regardless of the existence of a patent covering the product. Further, the invention, even if patented itself, cannot be commercialized if it infringes the valid patent rights of another party.
48
We rely on confidentiality agreements to protect our trade secrets. If these agreements are breached by our employees or other parties, our trade secrets may become known to our competitors.
We rely on trade secrets that we seek to protect through numerous measures, including non-compete and confidentiality agreements with our employees and other parties. If these agreements are breached, our competitors may obtain and use our trade secrets to gain a competitive advantage over us. Any remedies that may be available to us may not be adequate to protect our business or compensate us for the damaging disclosure. In addition, we may have to expend resources to protect our interests from possible infringement by others. For instance, we learned that a former employee, Qing Liang, Ph.D., who was employed by Actinium in the position of Vice President, Head of Radiation Sciences, violated the non-compete provision of her employment agreement by working for a direct competitor. Dr. Liang, who had access to materials containing proprietary information and trade secrets, is no longer employed by the direct competitor (who terminated her employment after learning of her actions). With the assistance of outside counsel and a forensic investigator, we also identified that Dr. Liang downloaded confidential information prior to the end of her employment with Actinium. We petitioned for and were granted a Stipulated Preliminary Injunction by the Supreme Court of the State of New York, New York County (Index No. 656841/2022) that ordered that Dr. Liang be enjoined from destroying or deleting any Actinium documents or information, enjoined from using, transmitting or transferring any Actinium Information (as defined in the injunction) other than to her counsel or Actinium’s counsel, ordered to return all Actinium information within 5 days of the Stipulated Preliminary Injunction, ordered to disclose to Actinium, under oath, all persons and devices to whom she transferred or disclosed Actinium Information, and ordered to allow a qualified forensic examiner selected by Actinium to remove and permanently delete all Actinium Information from any electronic devices, systems, email accounts, or other electronic or physical storage sites belonging to Dr. Liang. We also filed an arbitration proceeding against Dr. Liang and we intend to enforce the terms of her employment agreement, especially the non-compete and confidentiality provisions, to the fullest extent of our ability.
Risks Related to Our Operations
We expect to expand our development and regulatory capabilities and potentially implement sales, marketing and distribution capabilities, and, as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of product candidate development, regulatory affairs and, if any of our product candidates receives marketing approval, sales, marketing and distribution.
We currently do not have a marketing or sales team for the marketing, sales and distribution of any of our product candidates that are potentially able to obtain regulatory approval. In order to commercialize any product candidates, we must build on a territory-by-territory basis marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. If our product candidates receive regulatory approval, we intend to establish an internal sales or marketing team with technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be expensive and time consuming and will require significant attention of our executive officers to manage. We will also have to compete with other pharmaceutical and biotechnology companies to recruit, hire, train and retain marketing and sales personnel. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of any of our product candidates that we obtain approval to market.
To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources and the limited experience of our management team in managing a public company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
The use of hazardous materials, including radioactive and biological materials, in our research and development efforts imposes certain compliance costs on us and may subject us to liability for claims arising from the use or misuse of these materials.
Our research, development and manufacturing activities involve the controlled use of hazardous materials, including chemicals, radioactive and biological materials, such as radioactive isotopes. We are subject to federal, state, local and foreign environmental laws and regulations governing, among other matters, the handling, storage, use and disposal of these materials and some waste products. We cannot completely eliminate the risk of contamination or injury from these materials and we could be held liable for any damages that result, which could exceed our financial resources. We currently maintain insurance coverage for injuries resulting from the hazardous materials we use; however, future claims may exceed the amount of our coverage. Also, we do not have insurance coverage for pollution cleanup and removal. Currently the costs of complying with such federal, state, local and foreign environmental regulations are not significant, and consist primarily of waste disposal expenses. However, they could become expensive, and current or future environmental laws or regulations may impair our research, development, production and commercialization efforts.
49
We may undertake international operations, which will subject us to risks inherent with operations outside of the United States.
Although we do not have any international operations at this time, we intend to seek market clearances in foreign markets that we believe will generate significant opportunities. However, even with the cooperation of a commercialization partner, conducting drug development in foreign countries involves inherent risks, including, but not limited to difficulties in staffing, funding and managing foreign operations; unexpected changes in regulatory requirements; export restrictions; tariffs and other trade barriers; difficulties in protecting, acquiring, enforcing and litigating intellectual property rights; fluctuations in currency exchange rates; and potentially adverse tax consequences.
If we were to experience any of the difficulties listed above, or any other difficulties, any international development activities and our overall financial condition may suffer and cause us to reduce or discontinue our international development and registration efforts.
We are highly dependent on our key personnel, and if we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our future operations and successes depend in large part upon the continued service of key members of our senior management team whom we are highly dependent upon to manage our business. If any member of our current senior management terminates his or her employment with us and we are unable to find a suitable replacement quickly, the departure could have a material adverse effect on our business. An overall tightening and increasingly competitive labor market has been observed in the U.S. employment market generally, especially in response to the COVID-19 pandemic. Specific to the biotechnology industry in which we operate, there is significant demand and competition for highly specialized talent that we require. A sustained labor shortage or increased turnover rates within our employee base, caused by the COVID-19 pandemic, as a result of general macroeconomic factors, or due to dynamics within our industry, could lead to increased costs, such as increased wage rates to attract and retain employees, and could negatively affect our ability to efficiently conduct our clinical development, R&D, business development and potential regulatory and commercial activities. If we are unable to hire and retain employees capable of performing at a high-level, or if mitigation measures we may take to respond to a decrease in labor availability, have unintended negative effects, our business could be adversely affected. An overall labor shortage, lack of skilled labor, increased turnover or labor inflation, caused by the COVID-19 pandemic, general macroeconomic factors or as a result of biotechnology industry dynamics could have a material adverse impact on our operations, results of operations, liquidity or cash flows.
Our future success also depends on our ability to identify, attract, hire or engage, retain and motivate other well-qualified managerial, technical, clinical and regulatory personnel. This activity is likely to create additional demands on the time and attention of our senior management personnel as they identify, hire, and train external and internal candidates to fill the sizable number of positions required to execute our business plans, including submitting a BLA and building a commercial organization. The market for talent in our industry is very competitive. Many of the other biopharmaceutical companies we compete against for qualified personnel have greater financial and other resources, more favorable risk profiles and a longer operating history in the biopharmaceutical industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these opportunities may be more appealing to high-quality candidates than what we have to offer. There can be no assurance that such professionals will be available in the market, or that we will be able to retain existing professionals or meet or continue to meet their compensation requirements. Furthermore, the cost base in relation to such compensation, which may include equity compensation, may increase significantly, which could have a material adverse effect on us. Failure to establish and maintain an effective management team and workforce could adversely affect our ability to operate, grow and manage our business.
50
Managing our growth as we expand operations may strain our resources.
We expect to need to grow rapidly in order to support additional, larger, and potentially international, pivotal clinical trials of our product candidates as well as potential commercial operations, which will place a significant strain on our financial, managerial and operational resources. In order to achieve and manage growth effectively, we must continue to improve and expand our operational and financial management capabilities. Moreover, we will need to increase staffing and to train, motivate and manage our employees. All of these activities will increase our expenses and may require us to raise additional capital sooner than expected. Failure to manage growth effectively could materially harm our business, financial condition or results of operations.
We may expand our business through the acquisition of rights to new product candidates that could disrupt our business, harm our financial condition and may also dilute current stockholders’ ownership interests in our company.
Our business strategy includes expanding our products and capabilities, and we may seek acquisitions of product candidates, antibodies or technologies to do so. Acquisitions involve numerous risks, including substantial cash expenditures; potentially dilutive issuance of equity securities; incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition; difficulties in assimilating acquired technologies or the operations of the acquired companies; diverting our management’s attention away from other business concerns; risks of entering markets in which we have limited or no direct experience; and the potential loss of our key employees or key employees of the acquired companies.
We can make no assurances that any acquisition will result in short-term or long-term benefits to us. We may incorrectly judge the value or worth of an acquired product, company or business. In addition, our future success would depend in part on our ability to manage the rapid growth associated with some of these acquisitions. We cannot assure that we will be able to make the combination of our business with that of acquired products, businesses or companies work or be successful. Furthermore, the development or expansion of our business or any acquired products, business or companies may require a substantial capital investment by us. We may not have these necessary funds, or they might not be available to us on acceptable terms or at all. We may also seek to raise funds by selling shares of our preferred or common stock, which could dilute each current stockholder’s ownership interest in the Company.
Risks Related to Ownership of Our Common Stock
The sale of securities by us in any equity or debt financing could result in dilution to our existing stockholders and have a material adverse effect on our earnings.
We have financed our operations primarily through sales of stock and warrants. It is likely that during the next twelve months we will seek to raise additional capital through the sales of stock and warrants in order to expand our level of operations to continue our research and development efforts.
Any sale of common stock by us in a future offering could result in dilution to our existing stockholders as a direct result of our issuance of additional shares of our capital stock. In addition, our business strategy may include expansion through internal growth or by establishing strategic relationships with targeted customers and vendors. In order to do so, or to finance the cost of our other activities, we may issue additional equity securities that could dilute our stockholders’ stock ownership. We may also assume additional debt and incur impairment losses related to goodwill and other tangible assets if we acquire another company and this could negatively impact our earnings and results of operations.
51
Our common stock is subject to price volatility which could lead to losses by stockholders and potential costly security litigation.
The trading volume of our common stock has been and may continue to be extremely limited and sporadic. We expect the market price of our common stock to fluctuate substantially due to a variety of factors, including market perception of our ability to achieve our planned growth, quarterly operating results of other companies in the same industry, trading volume in our common stock, changes in general conditions in the economy and the financial markets or other developments affecting our competitors or us. This volatility has had a significant effect on the market price of securities issued by many companies for reasons unrelated to their operating performance and could have the same effect on our common stock.
The trading price of our common stock may be highly volatile and could fluctuate in response to factors such as:
| ● | actual or anticipated variations in our operating results; | |
| ● | announcements of developments by us or our competitors; | |
| ● | the timing of IND and/or BLA approval, the completion and/or results of our clinical trials; | |
| ● | regulatory actions regarding our products; | |
| ● | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; | |
| ● | adoption of new accounting standards affecting our industry; | |
| ● | additions or departures of key personnel; | |
| ● | introduction of new products by us or our competitors; | |
| ● | sales of our common stock or other securities in the open market; | |
| ● | inaccurate or unfavorable reports from securities or industry analysts; and | |
| ● | other events or factors, many of which are beyond our control. |
The stock market is subject to significant price and volume fluctuations. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been initiated against such a company. Litigation initiated against us, whether or not successful, could result in substantial costs and diversion of our management’s attention and our resources, which could harm our business and financial condition.
We do not intend to pay dividends on our common stock, so any returns will be determined by the value of our common stock.
We have never declared or paid any cash dividends on our common stock. For the foreseeable future, it is expected that earnings, if any, generated from our operations will be used to finance the growth of our business, and that no dividends will be paid to holders of our common stock. As a result, the success of an investment in our common stock will depend upon any future appreciation in its value. There is no guarantee that our common stock will appreciate in value.
52
Certain provisions of our Certificate of Incorporation and Bylaws and Delaware law make it more difficult for a third party to acquire us and make a takeover more difficult to complete, even if such a transaction were in our stockholders’ interest.
Provisions of our certificate of incorporation and bylaws may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our stock. Among other things, the certificate of incorporation and bylaws:
| ● | provide that the authorized number of directors may be changed by resolution of the board of directors; | |
| ● | provide that all vacancies, including newly-created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; | |
| ● | divide the board of directors into three classes; | |
| ● | provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner, and meet specific requirements as to the form and content of a stockholder’s notice; |
In addition, we are governed by Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. A “business combination” includes mergers, asset sales or other transactions resulting in a financial benefit to the stockholder. An “interested stockholder” is a person who, together with affiliates and associates, owns, or within three years, did own, 15% or more of the corporation’s outstanding voting stock. These provisions may have the effect of delaying, deferring or preventing a change in our control.
General Risk Factors
Compliance with the reporting requirements of federal securities laws can be expensive.
We are subject to the information and reporting requirements of the Exchange Act and other federal securities laws, and the compliance obligations of the Sarbanes-Oxley Act. The costs of preparing and filing annual and quarterly reports and other information with the Securities and Exchange Commission and furnishing audited reports to stockholders are substantial. In addition, we will incur substantial expenses in connection with the preparation of registration statements and related documents with respect any offerings of our common stock.
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited.
Our ability to utilize our federal net operating loss and tax credit carryforwards may be limited under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code. The limitations apply if we experience an “ownership change”, generally defined as a greater than 50 percentage point change in the ownership of our equity by certain stockholders over a rolling three-year period. Similar provisions of state tax law may also apply. We have not assessed whether such an ownership change has previously occurred. If we have experienced an ownership change at any time since our formation, we may already be subject to limitations on our ability to utilize our existing net operating losses and other tax attributes to offset taxable income. In addition, future changes in our stock ownership, which may be outside of our control, may trigger an ownership change and, consequently, the limitations under Sections 382 and 383 of the Code. As a result, if or when we earn net taxable income, our ability to use our pre-change net operating loss carryforwards and other tax attributes to offset such taxable income may be subject to limitations, which could adversely affect our future cash flows.
53
Failure to establish and maintain adequate finance infrastructure and accounting systems and controls could impair our ability to comply with the financial reporting and internal controls requirements for publicly traded companies.
As a public company, we operate in an increasingly demanding regulatory environment, including with respect to more complex accounting rules. Company responsibilities required by the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley Act, include establishing and maintaining corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud.
Our compliance with Section 404 of the Sarbanes-Oxley Act requires that we incur substantial accounting expense and expend significant management efforts. We complied with Section 404 at December 31, 2022 and 2021 and while our testing did not reveal any material weaknesses in our internal controls, any material weaknesses in our internal controls in the future would be required us to remediate in a timely manner so as to be able to comply with the requirements of Section 404 each year. If we are not able to comply with the requirements of Section 404 in a timely manner each year, we could be subject to sanctions or investigations by the SEC, NYSE American or other regulatory authorities which would require additional financial and management resources and could adversely affect the market price of our common stock. Furthermore, if we cannot provide reliable financial reports or prevent fraud, our business and results of operations could be harmed, and investors could lose confidence in our reported financial information.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, the price of our common stock and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. Multiple securities and industry analysts currently cover us. If one or more of the analysts downgrade our common stock or publish inaccurate or unfavorable research about our business, the price of our common stock would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for our common stock could decrease, which could cause the price of our common stock and trading volume to decline.
Our amended and restated bylaws, as amended, designate the U.S. federal district courts as the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act of 1933, as amended.
Our amended and restated bylaws, as amended, provide that, unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act of 1933, as amended. In addition, our amended and restated bylaws, as amended, state that any person purchasing or otherwise acquiring any interest in our security shall be deemed to have notice of and to have consented to such provision. Such choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits, if successful, might benefit our stockholders. Stockholders who do bring a claim in the federal district courts of the United States of America could face additional litigation costs in pursuing any such claim.
54
ITEM 1B. UNRESOLVED STAFF COMMENTS.
None.
ITEM 2. PROPERTIES.
We do not own any real property. We have leased offices at 275 Madison Avenue, New York, NY for seven years and our long-term lease for this space expired in 2022. We entered into a short-term lease for the same space until April 2023 with a monthly rate of $53 thousand. We are also responsible for certain other costs, such as insurance and maintenance. We issued a letter of credit of $391 thousand in connection with the lease and maintain a $391 thousand certified deposit as collateral for the letter of credit.
We entered into a lease for corporate office space at 100 Park Avenue, New York, NY effective June 1, 2022. The lease has a term of 5 years 2 months, with an expiration date in 2027, and a current annual rate of $599 thousand. We are also responsible for certain other costs, such as insurance, taxes, utilities and maintenance. We issued a letter of credit of $299 thousand in connection with the lease and maintain a $299 thousand certified deposit as collateral for the letter of credit.
We lease lab space and office space at Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY. The lease has a term of twelve months, expiring August 31, 2023, with a current annual rate of $136 thousand.
ITEM 3. LEGAL PROCEEDINGS.
From time to time, we may become involved in various lawsuits and legal proceedings, which arise in the ordinary course of business. Litigation is subject to inherent uncertainties, and an adverse result in these or other matters may arise from time to time that may harm business. We are currently not aware of any such legal proceedings or claims that will have, individually or in the aggregate, a material adverse effect on our business, financial condition or operating results.
ITEM 4. MINE SAFETY DISCLOSURES.
Not Applicable.
55
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDERS MATTERS, AND ISSUER PURCHASE OF EQUITY SECURITIES.
Market Information
Our common stock is listed for quotation on the NYSE American under the symbol “ATNM”.
Holders
As of March 31, 2023, there were 25,729,370 shares of common stock issued and outstanding, which were held by approximately 100 holders of record. There are no shares of preferred stock outstanding.
Dividends
We have never declared or paid any cash dividends on our common stock. For the foreseeable future, it is expected that earnings, if any, generated from our operations will be used to finance the growth of our business, and that no dividends will be paid to holders of our common stock. The decision to pay dividends is at the discretion of our Board of Directors and depends upon our financial condition, results of operations, capital requirements, and other factors that our Board of Directors deems relevant.
Securities Authorized for Issuance under Equity Compensation Plans
We currently have three equity compensation plans defined as follows:
The Company’s 2019 Amended and Restated Stock Plan has an expiration date of October 18, 2029 and the number of shares of our common stock authorized under the plan for grant to employees, directors and consultants is 9,333,333 shares.
The Company’s 2013 Amended and Restated Stock Plan has an expiration date of September 9, 2023 and after a number of amendments approved by stockholders, the number of shares of our common stock authorized under the plan for grant to employees, directors and consultants is 758,333 shares.
The Company’s 2013 Equity Incentive Plan has an expiration date of September 9, 2023 and the number of shares of our common stock authorized under the plan for grant to employees, directors and consultants under the plan is 33,333 shares.
The following table indicates shares of common stock authorized for issuance under our equity compensation plans as of December 31, 2022:
| Plan category | Number of securities to be issued upon exercise of outstanding options and restricted stock units (1) | Weighted- average exercise price of outstanding options and restricted stock units (2) | Number of securities remaining available for future issuance | |||||||||
| Equity compensation plans approved by security holders | 3,721,429 | $ | 8.00 | 6,383,638 | ||||||||
| Equity compensation plans not approved by security holders | - | - | - | |||||||||
| Total | 3,721,429 | $ | 8.00 | 6,383,638 | ||||||||
| (1) | Includes shares issuable upon the conversion of outstanding restricted stock units (“RSUs”). |
| (2) | The Weighted Average Exercise Price column does not include an amount for outstanding RSUs. |
ITEM 6. RESERVED.
56
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The information and financial data discussed below is derived from the audited consolidated financial statements of Actinium Pharmaceuticals, Inc. for its fiscal years ended December 31, 2022 and 2021. The consolidated financial statements of Actinium Pharmaceuticals, Inc. were prepared and presented in accordance with generally accepted accounting principles in the United States. The information and financial data discussed below is only a summary and should be read in conjunction with the historical financial statements and related notes of Actinium Pharmaceuticals, Inc. contained elsewhere in this Report. The financial statements contained elsewhere in this Report fully represent Actinium Pharmaceuticals, Inc.’s financial condition and operations; however, they are not indicative of the Company’s future performance. See “Cautionary Note Regarding Forward-Looking Statements” above for a discussion of forward-looking statements and the significance of such statements in the context of this Report.
Actinium Pharmaceuticals, Inc. is a clinical-stage, biopharmaceutical company applying its proprietary platform technology and clinical experience to develop novel targeted radiotherapies for patients with unmet needs. Our targeted radiotherapies combine the cell-killing ability of radiation via a radioisotope payload with a targeting agent, such as a monoclonal antibody, to deliver radiation in a precise manner inside the body to specific, targeted cells such as cancer cells, to potentially achieve greater efficacy with lower toxicity than with cytotoxic chemotherapy or external beam radiation. Targeted radiotherapies also enable broader application of radiation than external beam radiation as they can be used in the treatment of both solid tumors and blood cancers, which generally cannot be treated with external radiation given their diffuse nature.
Results of Operations – Year Ended December 31, 2022 Compared to the Year Ended December 31, 2021
The following table sets forth, for the periods indicated, data derived from our statements of operations:
| For the years ended December 31, | Increase | |||||||||||
| (amounts in thousands) | 2022 | 2021 | (Decrease) | |||||||||
| Revenue: | ||||||||||||
| Revenue | $ | - | $ | - | $ | - | ||||||
| Other revenue | 1,030 | 1,144 | (114 | ) | ||||||||
| Total revenue | 1,030 | 1,144 | (114 | ) | ||||||||
| Operating expenses: | ||||||||||||
| Research and development, net of reimbursements | 23,135 | 18,031 | 5,104 | |||||||||
| General and administrative | 11,999 | 8,077 | 3,922 | |||||||||
| Total operating expenses | 35,134 | 26,108 | 9,026 | |||||||||
| Other income | ||||||||||||
| Interest income – net | 1,087 | 190 | 897 | |||||||||
| Total other income | 1,087 | 190 | 897 | |||||||||
| Net loss | $ | (33,017 | ) | $ | (24,774 | ) | $ | (8,243 | ) | |||
Revenues
We recorded no commercial revenues for the years ended December 31, 2022 and 2021, respectively.
57
Other revenue
We determined that certain collaborations with a third-party are within the scope of Topic ASC 606, Revenue Recognition from Contracts with Customers, or ASC 606. The collaboration agreement is made up of multiple modules related to various research activities. While the third party has the option to terminate the agreement at the conclusion of any module, we identified a single performance obligation to provide research services within each module for which we receive monetary consideration. The consideration is recognized as revenue over each module and revenue of $0.9 million was recognized during each of the years ended December 31, 2022 and December 31, 2021.
The National Institutes of Health awarded us a Small Business Technology Transfer cost reimbursable grant to support a clinical collaboration with Memorial Sloan Kettering Cancer Center, or MSK, to study Iomab-ACT, our CD45-targeting Antibody Radio-Conjugate, for targeted conditioning to achieve lymphodepletion prior to administration of a CD19-targeted CAR T-cell therapy developed at MSK. We recognized other revenue during the years ended December 31, 2022 and 2021 of $0.1 million and $0.2 million, respectively, from this grant.
On April 7, 2022, we entered into a license and supply agreement with Immedica Pharma AB, or Immedica, pursuant to which Immedica licensed the exclusive product rights for commercialization of Iomab-B in the European Economic Area, Middle East and North Africa (EUMENA) including Algeria, Andorra, Bahrain, Cyprus, Egypt, Iran, Iraq, Israel, Jordan, Kuwait, Lebanon, Libya, Monaco, Morocco, Oman, Palestine, Qatar, San Marino, Saudi Arabia, Switzerland, Syria, Tunisia, Turkey, the United Arab Emirates, the United Kingdom, the Vatican City and Yemen. Upon signing, we were entitled to an upfront payment of $35 million from Immedica, which was received in May 2022. Under the terms of the License Agreement, we are eligible to receive regulatory and commercial milestone payments and are entitled to receive royalties in the mid-20 percent range on net sales of the product in certain countries that may result from the License Agreement. We will continue to be responsible for certain clinical development activities and the manufacturing of Iomab-B and will retain commercialization rights in the U.S. and rest of the world.
Our contract liabilities are recorded within Other revenue deferred – current liability or Long-term license revenue deferred in our condensed consolidated balance sheets depending on the short-term or long-term nature of the payments to be recognized. Our contract liabilities primarily consist of advanced payments from licensees. There was no Other revenue deferred-current liability at December 31, 2022, Other revenue deferred – current liability was $1.0 million at December 31, 2021. Long-term license revenue deferred was $35.0 million at December 31, 2022, resulting from the receipt from Immedica; there was no Long-term license revenue deferred at December 31, 2021. This deferred revenue will be recognized upon European Union regulatory approval of Iomab B.
Research and Development Expense, net of reimbursements
Research and development expenses increased by $5.1 million to $23.1 million for the year ended December 31, 2022 compared to $18.0 million for the year ended December 31, 2021. Higher expenses were primarily due to increased CMC activity related to Iomab-B, as well as increased compensation of $1.0 million resulting from increased headcount.
General and Administrative Expenses
General and administrative expenses increased by $3.9 million to $12.0 million for the year ended December 31, 2022 compared to $8.1 million for the year ended December 31, 2021. Higher expenses were primarily due to increased compensation of $0.9 million, increased non-cash equity compensation of $1.0 million, higher professional fees and consulting fees including recruitment costs, and higher legal fees.
Other Income
Other income is comprised of net interest income in both reporting periods. Other income of $1.1 million for the year ended December 31, 2022 increased from $0.2 million for the year ended December 31, 2021 due to a higher average balance and higher interest rates.
Net Loss
Net loss increased by $8.2 million to $33.0 million for the year ended December 31, 2022 compared to $24.8 million for the year ended December 31, 2021, primarily due to higher research and development expenses and general and administrative expenses, partially offset by other income.
58
Liquidity and Capital Resources
Historically, we have financed our operations primarily through sales of our common stock and common stock equivalents. The following tables sets forth selected cash flow information for the periods indicated:
| For the years ended December 31, | ||||||||
| (amounts in thousands) | 2022 | 2021 | ||||||
| Cash provided by/used in operating activities | $ | 8,644 | $ | (20,866 | ) | |||
| Cash used in investing activities | (366 | ) | (133 | ) | ||||
| Cash provided by financing activities | 23,109 | 35,221 | ||||||
| Net change in cash, cash equivalents and restricted cash | $ | 31,387 | $ | 14,222 | ||||
Net cash provided by operating activities for the year ended December 31, 2022 of $8.6 million increased by $29.5 million from a use of funds of $20.9 million for the year ended December 31, 2021. This increase was due to the receipt of the $35.0 million up-front payment from Immedica.
Net cash used in investing activities was $0.4 million and $0.1 million for the years ended December 31, 2022 and December 31, 2021, respectively, primarily due to the purchase of equipment for our laboratory space.
In August 2020 we entered into the Capital on Demand™ Sales Agreement with JonesTrading Institutional Services LLC, or JonesTrading, pursuant to which we would be able to sell, from time to time, through or to JonesTrading, up to an aggregate of $200 million of its common stock. On June 28, 2022, we entered into an Amendment and Restated Capital on Demand™ Sales Agreement, or the Amended Sales Agreement, with JonesTrading and B. Riley Securities, Inc. The Amended Sales Agreement modifies the original Capital on Demand™ Sales Agreement to include B. Riley as an additional sales agent thereunder. Shares of common stock are offered pursuant to a shelf registration statement on Form S-3 filed with the SEC on August 7, 2020. For the year ended December 31, 2022, we sold 3.5 million shares of common stock, resulting in gross proceeds of $23.9 million and net proceeds of $23.2 million. For the year ended December 31, 2021, we sold 4.6 million shares of common stock, resulting in gross proceeds of $36.5 million and net proceeds of $35.3 million. As of December 31, 2022, we have sold 10.2 million shares of common stock, resulting in gross proceeds of $83.0 million and net proceeds of $80.2 million relating to the Sales Agreement, as amended.
We entered into a lease for corporate office space effective June 1, 2022 and paid a security deposit to the landlord. The lease has a term of 5 years 2 months, with an expiration date in 2027, and current annual rent of $0.6 million. We are also responsible for certain other costs, such as insurance, utilities and maintenance. In July 2022, a certificate of deposit was provided as collateral for a letter of credit and the security deposit was returned.
We will require additional funds to conduct clinical and non-clinical trials, achieve regulatory approvals, and, subject to such approvals, commercially launch our product candidates, and will need to secure additional financing in the future to support our operations. As of the date of filing this report, we expect that our existing resources will be more than sufficient to fund our planned operations for more than 12 months following the date of this report. We base this belief on assumptions that are subject to change, and we may be required to use our available cash and cash equivalent resources sooner than we currently expect. Our actual future capital requirements will depend on many factors, including the progress and results of our ongoing clinical trials, the duration and cost of discovery and preclinical development, laboratory testing and clinical trials for our pipeline candidates, the timing and outcome of regulatory review of our product candidates, the costs involved in preparing, filing, prosecuting, maintaining, defending, and enforcing patent claims and other intellectual property rights, the number and development requirements of other pipeline candidates that we pursue, and the costs of commercialization activities, including product marketing, sales, and distribution.
We expect to continue to operate at a net loss as we continue our research and development efforts, continue to conduct clinical trials and develop manufacturing, sales, marketing and distribution capabilities. There can be no assurance that the products under development by us will be approved for sale in the United States or elsewhere. Our ability to obtain additional capital may depend on prevailing economic conditions and financial, business, and other factors beyond our control. Current economic conditions have been, and continue to be, volatile. Continued instability in these market conditions may limit our ability to access the capital necessary to fund and grow our business.
59
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
Critical Accounting Policies
Our management’s discussion and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and the disclosure of contingent assets and liabilities in our consolidated financial statements during the reporting periods. These items are monitored and analyzed by us for changes in facts and circumstances, and material changes in these estimates could occur in the future. We base our estimates on historical experience, known trends and events, and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Changes in estimates are reflected in reported results for the period in which they become known. Actual results may differ materially from these estimates under different assumptions or conditions
Fair Value Measurement
Fair value is defined as the price that would be received to sell an asset, or paid to transfer a liability, in an orderly transaction between market participants. A fair value hierarchy has been established for valuation inputs that gives the highest priority to quoted prices in active markets for identical assets or liabilities and the lowest priority to unobservable inputs.
Revenue Recognition
We recognize revenue in accordance with ASC 606. Under ASC 606, we recognize revenue when our customer obtains control of promised goods or services, in an amount that reflects the consideration that we expect to receive in exchange for those goods or services. To determine revenue recognition for arrangements within the scope of ASC 606, we perform the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price, including variable consideration, if any; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue as we satisfy a performance obligation. We only apply the five-step model to contracts when it is probable that we will collect the consideration to which we are entitled in exchange for the goods or services we transfer to the customer.
At contract inception, once the contract is determined to be within the scope of ASC 606, we assess whether the promised goods or services promised within each contract are distinct and, therefore, represent a separate performance obligation. Goods and services that are determined not to be distinct are combined with other promised goods and services until a distinct bundle is identified. In determining whether goods or services are distinct, we evaluate certain criteria, including whether (i) the customer can benefit from the good or service either on its own or together with other resources that are readily available to the customer (capable of being distinct) and (ii) the good or service is separately identifiable from other goods or services in the contract (distinct in the context of the contract).
ASC 606 requires us to allocate the arrangement consideration on a relative standalone selling price basis for each performance obligation after determining the transaction price of the contract and identifying the performance obligations to which that amount should be allocated. The relative standalone selling price is defined in the new revenue standard as the price at which an entity would sell a promised good or service separately to a customer. We then recognize as revenue the amount of the transaction price that is allocated to the respective performance obligation as each performance obligation is satisfied, either at a point in time or over time, and if over time, recognition is based on the use of an output or input method.
60
Collaborative Arrangements
We follow the accounting guidance for collaboration agreements, which requires that certain transactions between us and collaborators be recorded in our consolidated statements of operations on either a gross basis or net basis, depending on the characteristics of the collaborative relationship, and requires enhanced disclosure of collaborative relationships. We evaluate our collaboration agreements for proper classification in our consolidated statements of operations based on the nature of the underlying activity. When we conclude that we have a customer relationship with one of our collaborators, we follow the guidance of ASC 606.
Grant Revenue
We had a grant from a government-sponsored entity for research and development related activities that provided for payments for reimbursed costs, which included overhead and general and administrative costs as well as an administrative fee. We recognized revenue from the grant as we performed services under this arrangement. Associated expenses were recognized when incurred as research and development expense. Revenue and related expenses are presented gross in the consolidated statements of operations.
License Revenue
We entered into a product licensing agreement whereby we allowed a third party to commercialize a certain product in specified territories using our trademarks. The terms of this arrangement includes payment to us for a combination of one or more of the following: upfront license fees; development, regulatory and sales-based milestone payments; and royalties on net sales of licensed products. We use judgment to determine whether milestones or other variable consideration should be included in the transaction price.
Upfront license fees: If the license to our intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, we will recognize revenue from upfront license fees allocated to the license when the license is transferred to the licensee and the licensee is able to use and benefit from the license. For licenses that are bundled with other promises, we determine whether the combined performance obligation is satisfied over time or at a point in time.
Development, regulatory or commercial milestone payments: At the inception of each arrangement that includes payments based on the achievement of certain development, regulatory and sales-based or commercial events, we evaluate whether the milestones are considered probable of being achieved and estimate the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within our or the licensee’s control, such as regulatory approvals, are not considered probable of being achieved until regulatory approval is received. At the end of each subsequent reporting period, we will re-evaluate the probability of achieving such development and regulatory milestones and any related constraint, and if necessary, adjust our estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis and recorded as part of license revenues during the period of adjustment.
Sales-based milestone payments and royalties: For arrangements that include sales-based royalties, including milestone payments based on the volume of sales, we will determine whether the license is deemed to be the predominant item to which the royalties or sales-based milestones relate and if such is the case, we will recognize revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied).
Upfront payments and fees may require deferral of revenue recognition to a future period until we perform our obligations under these arrangements or when it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with any variable consideration is subsequently resolved. Amounts payable to us are recorded as accounts receivable when our right to consideration is unconditional.
61
Research and Development Costs
Research and development costs are expensed as incurred. These costs include the costs of manufacturing drug components and final drug product, the costs of clinical trials, costs of employees and associated overhead, and depreciation and amortization costs related to facilities and equipment. Research and development reimbursements are recorded by us as a reduction of research and development costs.
Share-Based Payments
We estimate the fair value of each stock option award at the grant date by using the Black-Scholes option pricing model. The fair value determined represents the cost for the award and is recognized over the vesting period during which an employee is required to provide service in exchange for the award. We account for forfeitures of stock options as they occur.
Income Taxes
We use the asset and liability method to calculate deferred taxes. Deferred taxes are recognized based on the differences between the financial reporting and income tax bases of assets and liabilities using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. We review deferred tax assets for a valuation allowance based upon whether it is more likely than not that the deferred tax asset will be fully realized. A valuation allowance, if necessary, is provided against deferred tax assets, based upon our assessment as to their realization.
We recognize tax when the positions meet a “more-likely-than-not” recognition threshold. There were no tax positions for which it is considered reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within the next year. We recognize interest related to unrecognized tax benefits in interest expense and penalties in operating expenses.
Accounting Standards Recently Adopted
In May 2021, the Financial Accounting Standards Board, or FASB, issued ASU 2021-04, Earnings Per Share (topic 260), Debt — Modifications and Extinguishments (Subtopic 470-50), Compensation – Stock Compensation (Topic 718) and Derivatives and Hedging – Contracts in an Entity’s Own Equity (Subtopic 815-40) – Issuer’s Accounting for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options, which provides guidance of a modification or an exchange of a freestanding equity-classified written call option that remains equity classified after modification or exchange as (1) an adjustment to equity and, if so, the related earnings per share (EPS) effects, if any, or (2) an expense and, if so, the manner and pattern of recognition. The amendments in this ASU are effective January 1, 2022, including interim periods. We adopted this standard effective January 1, 2022 and the standard did not have a material effect on our financial statements.
In November 2021, the FASB issued ASU 2021-10, Government Assistance (Topic 832), Disclosures by Business Entities about Government Assistance, which provides guidance on disclosure requirements to entities other than not-for-profit entities about transaction with a government that are accounted for by applying a grant or contribution accounting model by analogy. ASU 2021-10 requires an entity to make annual disclosures related to (1) the nature of the transactions and the related accounting policy used to account for the government transactions, (2) quantification and disclosure of amounts related to the government transactions included in balance sheet and income statement financial statement line items, and (3) significant terms and conditions of the government transactions, including commitments and contingencies. The amendments of ASU 2021-10 are effective January 1, 2022, including interim periods. We adopted this standard effective January 1, 2022 and the standard did not have a material impact on our financial statements.
62
Accounting Standards Recently Issued
In October 2021, FASB issued ASU 2021-08, Business Combinations (Topic 805), Account for Contract Assets and Contract Liabilities from Contracts with Customers, which provides guidance on accounting for contract assets and contract liabilities acquired in a business combination in accordance with ASC 606. To achieve this, an acquirer may assess how the acquiree applied ASC 606 to determine what to record for the acquired revenue contracts. Generally, this should result in an acquirer recognizing and measuring the acquired contract assets and contract liabilities consistent with how they were recognized and measured in the acquiree’s financial statements. The amendments of ASU 2021-08 are effective January 1, 2023, including interim periods. Early adoption is permitted, including adoption in an interim period. We will evaluate the impact of ASU 2021-08 on any future business combinations that we may enter in the future.
Subsequent Event
Since December 31, 2022, we have sold 0.1 million shares of common stock under our Amended Sales Agreement, resulting in net proceeds of $0.8 million.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
We are not currently exposed to significant market risk related to changes in interest rates. As of December 31, 2022, our cash equivalents consisted primarily of short-term money market funds. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates. Due to the short-term nature of the cash equivalents in our portfolio and the low risk profile of our cash equivalents, an immediate 10% change in interest rates would not have a material effect on the fair market value of our financial position or results of operations.
We are not currently exposed to significant market risk related to changes in foreign currency exchange rates. Our operations may be subject to fluctuations in foreign currency exchange rates in the future.
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not believe that inflation had a material effect on our business, financial condition, or results of operations during the years ended December 31, 2022 and 2021.
63
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of
Actinium Pharmaceuticals, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Actinium Pharmaceuticals, Inc. (the “Company”) as of December 31, 2022 and 2021, the related consolidated statements of operations, changes in stockholders’ equity and cash flows for each of the two years in the period ended December 31, 2022 and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2022 and 2021, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2022, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
/s/ Marcum llp
We have served as the Company’s auditor since 2012.
March 31, 2023
F-1
Actinium Pharmaceuticals, Inc.
Consolidated Balance Sheets
(amounts in thousands, except share and per share data)
| December 31, 2022 | December 31, 2021 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Restricted cash – current | ||||||||
| Security deposit | ||||||||
| Prepaid expenses and other current assets | ||||||||
| Total Current Assets | ||||||||
| Property and equipment, net of accumulated depreciation of $ | ||||||||
| Restricted cash – long term | ||||||||
| Operating lease right-of-use assets | ||||||||
| Finance leases right-of-use assets | ||||||||
| Total Assets | $ | $ | ||||||
| Liabilities and Stockholders’ Equity | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable and accrued expenses | $ | $ | ||||||
| Other revenue deferred – current liability | ||||||||
| Operating leases current liability | ||||||||
| Finance leases current liability | ||||||||
| Total Current Liabilities | ||||||||
| Long-term license revenue deferred | ||||||||
| Long-term operating lease obligations | ||||||||
| Long-term finance lease obligations | ||||||||
| Total Liabilities | ||||||||
| Commitments and contingencies | ||||||||
| Stockholders’ Equity: | ||||||||
| Preferred stock, $ | ||||||||
| Common stock, $ | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total Stockholders’ Equity | ||||||||
| Total Liabilities and Stockholders’ Equity | $ | $ | ||||||
See accompanying notes to the consolidated financial statements.
F-2
Actinium Pharmaceuticals, Inc.
Consolidated Statements of Operations
(amounts in thousands, except share and per share data)
| For the Year ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Revenue | ||||||||
| Revenue | $ | $ | ||||||
| Other Revenue | ||||||||
| Total revenue | ||||||||
| Operating expenses: | ||||||||
| Research and development, net of reimbursements | ||||||||
| General and administrative | ||||||||
| Total operating expenses | ||||||||
| Loss from operations | ( | ) | ( | ) | ||||
| Other income: | ||||||||
| Interest income - net | ||||||||
| Total other income | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| $ | ( | ) | $ | ( | ) | |||
See accompanying notes to the consolidated financial statements.
F-3
Actinium Pharmaceuticals, Inc.
Consolidated Statements of Changes in Stockholders’ Equity
For the Years Ended December 31, 2022 and 2021
(amounts in thousands, except share amounts)
| Common Stock | Additional Paid-In | Accumulated | Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | Deficit | Equity | ||||||||||||||||
| Balance, January 1, 2021 | $ | $ | $ | ( | ) | $ | ||||||||||||||
| Stock-based compensation | ||||||||||||||||||||
| Sale of common stock, net of offering costs | ||||||||||||||||||||
| Issuance of common stock from exercise of stock options | ||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance, December 31, 2021 | $ | $ | $ | ( | ) | $ | ||||||||||||||
| Stock-based compensation | ||||||||||||||||||||
| Sale of common stock, net of offering costs | ||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance, December 31, 2022 | $ | $ | $ | ( | ) | $ | ||||||||||||||
See accompanying notes to the consolidated financial statements.
F-4
Actinium Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(amounts in thousands)
| For the Year ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Cash Flows from Operating Activities: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation expense | ||||||||
| Depreciation and amortization expense | ||||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | ( | ) | ( | ) | ||||
| Accounts payable and accrued expenses | ||||||||
| Other revenue deferred – current liability | ( | ) | ||||||
| Long-term license revenue deferred | ||||||||
| Operating lease liabilities | ( | ) | ( | ) | ||||
| Net Cash Provided By/Used In Operating Activities | ( | ) | ||||||
| Cash Flows Used in Investing Activities: | ||||||||
| Purchase of property and equipment | ( | ) | ( | ) | ||||
| Net Cash Used In Investing Activities | ( | ) | ( | ) | ||||
| Cash Flows from Financing Activities: | ||||||||
| Payments on finance leases | ( | ) | ( | ) | ||||
| Proceeds from sales of shares of common stock, net of offering costs | ||||||||
| Proceeds from the exercise of stock options | ||||||||
| Net Cash Provided By Financing Activities | ||||||||
| Net change in cash, cash equivalents and restricted cash | ||||||||
| Cash, cash equivalents and restricted cash at beginning of year | ||||||||
| Cash, cash equivalents and restricted cash at end of year | $ | $ | ||||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for interest | $ | $ | ||||||
| Cash paid for taxes | $ | $ | ||||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Right-of-use assets obtained in exchange for lease liabilities | $ | $ | ||||||
| Equipment obtained in exchange for security deposit | $ | $ | ||||||
See accompanying notes to the consolidated financial statements.
F-5
Actinium Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
Note 1 - Description of Business and Summary of Significant Accounting Policies
Nature of Business - Actinium Pharmaceuticals, Inc. is a biopharmaceutical company developing targeted radiotherapies to deliver cancer-killing radiation with cellular level precision to treat patients with high unmet medical needs.
Principles of Consolidation - The consolidated financial statements include the Company’s accounts and those of the Company’s wholly owned subsidiaries. All significant intercompany accounts and transactions have been eliminated.
Use of Estimates in Financial Statement Presentation - The preparation of these consolidated financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the consolidated financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents and Restricted Cash- The Company considers all highly liquid accounts with original maturities of three months or less to be cash equivalents. Balances held by the Company are typically in excess of Federal Deposit Insurance Corporation insured limits.
Following is a summary of cash, cash equivalents and restricted cash at December 31, 2022 and December 31, 2021:
| (in thousands) | December 31, 2022 | December 31, 2021 | ||||||
| Cash and cash equivalents | $ | $ | ||||||
| Restricted cash – current | ||||||||
| Restricted cash – long-term | ||||||||
| Cash, cash equivalents and restricted cash | $ | $ | ||||||
Restricted cash relates to certificates of deposit held as collateral for letters of credit issued in connection with the Company’s leases of corporate office spaces.
Property and Equipment - Machinery and equipment are recorded at cost and depreciated on a straight-line basis over estimated useful lives of three to five years. Furniture and fixtures are recorded at cost and depreciated on a straight-line basis over estimated useful lives of seven years. When assets are retired, the cost and related accumulated depreciation are removed from the accounts, and any related gain or loss is reflected in operations. Repairs and maintenance expenditures are charged to operations. Capitalized lease assets are recorded at the lesser of the present value of minimum lease payments or fair value and amortized over the estimated useful life of the related property or term of the lease.
Leases – The Company has operating and finance leases for corporate office space and office equipment located at the corporate office space. Leases with an initial term of 12 months or less are not recorded on the balance sheet; lease expense for these leases is recognized on a straight-line basis over the lease term. The Company entered into a lease for corporate office space effective June 1, 2022 and paid a security deposit to the landlord. A certificate of deposit was provided as collateral for a letter of credit issued with this office space during 2022 and at that time, the security deposit was returned to the Company.
F-6
Fair Value Measurement - Fair value is defined as the price that would be received to sell an asset, or paid to transfer a liability, in an orderly transaction between market participants. A fair value hierarchy has been established for valuation inputs that gives the highest priority to quoted prices in active markets for identical assets or liabilities and the lowest priority to unobservable inputs.
Revenue Recognition - The Company recognizes revenue in accordance with Accounting Standards Codification (ASC) Topic 606, Revenue From Contracts With Customers (“ASC 606”). Under ASC 606, an entity recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the consideration that the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements within the scope of ASC 606, the entity performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price, including variable consideration, if any; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue as the entity satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the entity will collect the consideration to which it is entitled in exchange for the goods or services it transfers to the customer.
At contract inception, once the contract is determined to be within the scope of ASC 606, the Company assesses whether the promised goods or services promised within each contract are distinct and, therefore, represent a separate performance obligation. Goods and services that are determined not to be distinct are combined with other promised goods and services until a distinct bundle is identified. In determining whether goods or services are distinct, the Company evaluates certain criteria, including whether (i) the customer can benefit from the good or service either on its own or together with other resources that are readily available to the customer (capable of being distinct) and (ii) the good or service is separately identifiable from other goods or services in the contract (distinct in the context of the contract).
The Company then determines the transaction price, which is the amount of consideration it expects to be entitled from a customer in exchange for the promised goods or services for each performance obligation and recognizes the associated revenue as each performance obligation is satisfied. The Company’s estimate of the transaction price for each contract includes all variable consideration to which it expects to be entitled. Variable consideration includes payments in the form of collaboration milestone payments. If an arrangement includes collaboration milestone payments, the Company evaluates whether the milestones are considered probable of being reached and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price.
ASC 606 requires the Company to allocate the arrangement consideration on a relative standalone selling price basis for each performance obligation after determining the transaction price of the contract and identifying the performance obligations to which that amount should be allocated. The relative standalone selling price is defined in the revenue standard as the price at which an entity would sell a promised good or service separately to a customer. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation as each performance obligation is satisfied, either at a point in time or over time, and if over time, recognition is based on the use of an output or input method.
Collaborative Arrangements - The Company follows the accounting guidance for collaboration agreements with third parties, which requires that certain transactions between the Company and collaborators be recorded in its consolidated statements of operations on either a gross basis or net basis, depending on the characteristics of the collaborative relationship, and requires enhanced disclosure of collaborative relationships. The Company evaluates its collaboration agreements for proper classification in its consolidated statements of operations based on the nature of the underlying activity. When the Company has concluded that it has a customer relationship with one of its collaborators, the Company follows the guidance of ASC 606.
Grant Revenue – The Company had a grant from a government-sponsored entity for research and development related activities that provided for payments for reimbursed costs, which included overhead and general and administrative costs as well as an administrative fee. The Company recognized revenue from grants as it performed services under this arrangement. Associated expenses were recognized when incurred as research and development expense. Revenue and related expenses are presented gross in the consolidated statements of operations.
F-7
License Revenue – The Company entered into a product licensing agreement whereby the Company allowed a third party to commercialize a certain product in specified territories using the Company’s trademarks. The terms of this arrangement includes payment to the Company for a combination of one or more of the following: upfront license fees; development, regulatory and sales-based milestone payments; and royalties on net sales of licensed products. The Company uses its judgment to determine whether milestones or other variable consideration should be included in the transaction price.
Upfront license fees: If the license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, the Company will recognize revenue from upfront license fees allocated to the license when the license is transferred to the licensee and the licensee is able to use and benefit from the license. For licenses that are bundled with other promises, the Company determines whether the combined performance obligation is satisfied over time or at a point in time.
Development, regulatory or commercial milestone payments: At the inception of each arrangement that includes payments based on the achievement of certain development, regulatory and sales-based or commercial events, the Company evaluates whether the milestones are considered probable of being achieved and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within the Company’s or the licensee’s control, such as regulatory approvals, are not considered probable of being achieved until regulatory approval is received. At the end of each subsequent reporting period, the Company will re-evaluate the probability of achieving such development and regulatory milestones and any related constraint, and if necessary, adjust the Company’s estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis and recorded as part of license revenue during the period of adjustment.
Sales-based milestone payments and royalties: For arrangements that include sales-based royalties, including milestone payments based on the volume of sales, the Company will determine whether the license is deemed to be the predominant item to which the royalties or sales-based milestones relate and if such is the case, the Company will recognize revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied).
Upfront payments and fees may require deferral of revenue recognition to a future period until the Company performs its obligations under these arrangements or when it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur or when the uncertainty associated with any variable consideration is subsequently resolved. Amounts payable to the Company are recorded as accounts receivable when the Company’s right to consideration is unconditional.
Research and Development Costs - Research and development costs are expensed as incurred. These costs include the costs of manufacturing drug product, the costs of clinical trials, costs of employees and associated overhead, and depreciation and amortization costs related to facilities and equipment. Research and development reimbursements are recorded by the Company as a reduction of research and development costs.
Share-Based Payments - The Company estimates the fair value of each stock option award at the grant date by using the Black-Scholes option pricing model. The fair value determined represents the cost for the award and is recognized over the vesting period during which an employee is required to provide service in exchange for the award. The Company accounts for forfeitures of stock options as they occur.
Income Taxes - The Company accounts for income taxes in accordance with ASC 740 Income Taxes, which requires the asset and liability method to calculate deferred taxes. Deferred taxes are recognized based on the differences between the financial reporting and income tax bases of assets and liabilities using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company reviews deferred tax assets for a valuation allowance based upon whether it is more likely than not that the deferred tax asset will be fully realized.
ASC 740 prescribes guidance for the financial statement recognition, measurement and disclosure of uncertain tax positions. Tax positions must meet a “more-likely-than-not” recognition threshold to be recognized. There were no tax positions for which it is considered reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within the next year. The Company recognizes interest related to unrecognized tax benefits in interest expense and penalties in operating expenses
F-8
Net Loss Per Common Share - Basic loss per common share is computed by dividing the net loss available to common stockholders by the weighted average number of common shares outstanding during the reporting period. For periods of net loss, diluted loss per share is calculated similarly to basic loss per share because the impact of all potential dilutive common shares is anti-dilutive.
For the years ended December 31, 2022 and 2021, the Company’s potentially dilutive shares, which include outstanding common stock options, restricted stock units and warrants, have not been included in the computation of diluted net loss per share as the result would have been anti-dilutive.
| (in thousands) | December 31, 2022 | December 31, 2021 | ||||||
| Stock Options | ||||||||
| Restricted Stock Units | ||||||||
| Warrants | ||||||||
| Total | ||||||||
Subsequent Events - The Company’s management reviewed all material events through the date the consolidated financial statements were issued for subsequent event disclosure consideration.
Recently Adopted Accounting Pronouncements – In May 2021, FASB issued ASU 2021-04, Earnings Per Share (topic 260), Debt — Modifications and Extinguishments (Subtopic 470-50), Compensation – Stock Compensation (Topic 718) and Derivatives and Hedging – Contracts in an Entity’s Own Equity (Subtopic 815-40) – Issuer’s Accounting for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options, which provides guidance of a modification or an exchange of a freestanding equity-classified written call option that remains equity classified after modification or exchange as (1) an adjustment to equity and, if so, the related earnings per share (EPS) effects, if any, or (2) an expense and, if so, the manner and pattern of recognition. The amendments in this ASU are effective January 1, 2022, including interim periods. The Company adopted this standard effective January 1, 2022 and the standard did not have a material effect on the Company’s financial statements.
In November 2021, the FASB issued ASU 2021-10, Government Assistance (Topic 832), Disclosures by Business Entities about Government Assistance, which provides guidance on disclosure requirements to entities other than not-for-profit entities about transaction with a government that are accounted for by applying a grant or contribution accounting model by analogy. ASU 2021-10 requires an entity to make annual disclosures related to (1) the nature of the transactions and the related accounting policy used to account for the government transactions, (2) quantification and disclosure of amounts related to the government transactions included in balance sheet and income statement financial statement line items, and (3) significant terms and conditions of the government transactions, including commitments and contingencies. The amendments of ASU 2021-10 are effective January 1, 2022, including interim periods. The Company adopted this standard effective January 1, 2022, and the standard did not have a material impact on the Company’s financial statements.
F-9
Recently Issued Accounting Pronouncements – In October 2021, FASB issued ASU 2021-08, Business Combinations (Topic 805), Account for Contract Assets and Contract Liabilities from Contracts with Customers, which provides guidance on accounting for contract assets and contract liabilities acquired in a business combination in accordance with ASC 606. To achieve this, an acquirer may assess how the acquiree applied ASC 606 to determine what to record for the acquired revenue contracts. Generally, this should result in an acquirer recognizing and measuring the acquired contract assets and contract liabilities consistent with how they were recognized and measured in the acquiree’s financial statements. The amendments of ASU 2021-08 are effective January 1, 2023, including interim periods. Early adoption is permitted, including adoption in an interim period. The Company will evaluate the impact of ASU 2021-08 on any future business combinations the Company may enter in the future.
Note 2 - Prepaid Expenses and Other Current Assets
Prepaid expenses and other current assets consisted of the following at December 31, 2022 and 2021:
| December 31, | December 31, | |||||||
| 2022 | 2021 | |||||||
| Prepaid insurance | $ | $ | ||||||
| Prepaid clinical trial expenses | ||||||||
| Other prepaid expenses and other current assets | ||||||||
| Total prepaid expenses and other current assets | $ | $ | ||||||
Note 3 - Property and Equipment
Property and equipment consisted of the following at December 31, 2022 and 2021:
| December 31, | December 31, | |||||||||||
| (in thousands) | Lives | 2022 | 2021 | |||||||||
| Lab equipment | $ | $ | ||||||||||
| Office equipment and furniture | ||||||||||||
| Less: accumulated depreciation | ( | ) | ( | ) | ||||||||
| Property and equipment, net | $ | $ | ||||||||||
Depreciation expense consisted of the following for the years ended December 31, 2022 and 2021, respectively:
| December 31, | December 31, | |||||||
| (in thousands) | 2022 | 2021 | ||||||
| Research and development | $ | $ | ||||||
| General administrative | ||||||||
| Total Depreciation expense | $ | $ | ||||||
Note 4 - Leases
The Company determines if an arrangement is a lease at inception. This determination generally depends on whether the arrangement conveys to the Company the right to control the use of a fixed asset for a period of time in exchange for consideration. Control of an underlying asset is conveyed to the Company if the Company obtains the rights to direct the use of and to obtain substantially all of the economic benefits from using the underlying asset. The Company has lease agreements which include lease and non-lease components, which the Company has elected to account for as a single lease component for all classes of underlying assets. Lease expense for variable lease components are recognized when the obligation is probable. The Company made an accounting policy election to exclude from balance sheet reporting those leases with initial terms of 12 months or less.
Right-of-use assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term. ASC 842 requires a lessee to discount its unpaid lease payments using the interest rate implicit in the lease or, if that rate cannot be readily determined, its incremental borrowing rate. As an implicit interest rate was not readily determinable in the Company’s leases, the incremental borrowing rate was used based on the information available at commencement date in determining the present value of lease payments.
F-10
The lease term for all of the Company’s leases includes the non-cancellable period of the lease plus any additional periods covered by either a Company option to extend (or not to terminate) the lease that the Company is reasonably certain to exercise, or an option to extend (or not to terminate) the lease controlled by the lessor. Options for lease renewals have been excluded from the lease term (and lease liability) for the majority of the Company’s leases as the reasonably certain threshold is not met.
The Company entered into a lease
for corporate office space, effective June 1, 2022. The lease has a term of
At December 31, 2021, for capitalization purposes under ASC842, the Company had an operating lease for corporate office space that expired in 2022 and finance leases for office equipment and furniture located in the corporate office space. In addition, the Company has auxiliary corporate office space that it rents on a month-to-month basis; this rental was accounted for as an operating lease with the same term as the Company’s office space.
The components of lease expense are as follows:
| (in thousands) | Year ended December 31, 2022 | Year ended December 31, 2021 | ||||||
| Operating lease expense | $ | $ | ||||||
| Finance lease cost | ||||||||
| Amortization of right-to-use assets | $ | $ | ||||||
| Interest on lease liabilities | $ | $ | ||||||
| Total finance lease cost | $ | |||||||
Supplemental cash flow information related to leases are as follows:
| Year ended | ||||||||
| (in thousands) | December 31, 2022 | December 31, 2021 | ||||||
| Cash flow information: | ||||||||
| Cash paid for amounts included in the measurement of lease liabilities: | ||||||||
| Operating cash flow use from operating leases | $ | $ | ||||||
| Operating cash flow use from finance leases | $ | $ | ||||||
| Financing cash flow use from finance leases | $ | $ | ||||||
| Non-cash activity: | ||||||||
| Right-of-use assets obtained in exchange for lease obligations: | ||||||||
| Operating leases | $ | $ | ||||||
| Finance Leases | $ | $ | ||||||
Weighted average remaining lease terms are as follows at December 31, 2022:
| Weighted average remaining lease term: | ||
| Operating leases | ||
| Finance Leases |
F-11
As the interest rate implicit in the leases was not readily determinable at the time that the leases were evaluated, the Company used its incremental borrowing rate based on the information available in determining the present value of lease payments. The Company’s incremental borrowing rate was based on the term of the lease, the economic environment of the lease and reflect the rate the Company would have had to pay to borrow on a secured basis. Below is information on the weighted average discount rates used at the time that the leases were evaluated:
| Weighted average discount rates: | ||||
| Operating leases | % | |||
| Finance Leases | % | |||
Maturities of lease liabilities are as follows:
| Year ending December 31, | Operating Leases | Finance Leases | ||||||
| 2023 | ||||||||
| 2024 | ||||||||
| 2025 | ||||||||
| 2026 | ||||||||
| 2027 | ||||||||
| Total lease payments | $ | $ | ||||||
| Less imputed interest | ( | ) | ||||||
| Present value of lease liabilities | $ | $ | ||||||
Note 5 - Other revenue
The Company determined that
certain collaborations with a third party are within the scope of ASC 606. The collaboration agreement is made up of multiple modules
related to various research activities. The Company identified a single performance obligation to provide research services within each
module for which the Company receives monetary consideration. The third party can choose to proceed with each module or can terminate
the agreement at any time. The Company recognizes revenue for each module on a straight-line basis over the expected module period. Revenue
for succeeding modules is not recognized until all contingencies are resolved, inclusive of the third party’s ability to terminate
the module. The consideration is recognized to revenue over each module and revenue of $
The Company had a grant from
a government-sponsored entity for research and development related activities that provide for payments for reimbursed costs, which included
overhead and general and administrative costs as well as an administrative fee. The Company recognized revenue from grants as it performed
services under this arrangement. Associated expenses are recognized when incurred as research and development expense. Other revenue recognized
from this grant during the years ended December 31, 2022 and 2021 was $
On April 7, 2022, the Company
entered into a license and supply agreement (the “License Agreement”) with Immedica Pharma AB (“Immedica”), pursuant
to which Immedica licensed the exclusive product rights for commercialization of Iomab-B (I-131 apamistamab) in the European Economic
Area, Middle East and North Africa (EUMENA) including Algeria, Andorra, Bahrain, Cyprus, Egypt, Iran, Iraq, Israel, Jordan, Kuwait, Lebanon,
Libya, Monaco, Morocco, Oman, Palestine, Qatar, San Marino, Saudi Arabia, Switzerland, Syria, Tunisia, Turkey, the United Arab Emirates,
the United Kingdom, the Vatican City and Yemen. Upon signing, the Company was entitled to an upfront payment of $
The Company’s contract
liabilities are recorded within Other revenue deferred – current liability or Long-term license revenue deferred in its condensed
consolidated balance sheets, depending on the short-term or long-term nature of the payments to be recognized. The Company’s contract
liabilities primarily consist of advanced payments from licensees. There was no Other revenue deferred – current liability at December
31, 2022 and $
F-12
Note 6 - Commitments and Contingencies
On June 15, 2012, the Company
entered into a license and sponsored research agreement with Fred Hutchinson Cancer Research Center (“FHCRC”) to build upon
previous and ongoing clinical trials with apamistamab (licensed antibody). FHCRC has completed both a Phase 1 and Phase 2 clinical trial
with apamistamab. The Company has been granted exclusive rights to the antibody and related master cell bank developed by FHCRC. A milestone
payment of $
Note 7 - Equity
In August 2020, the Company entered
into the Capital on Demand™ Sales Agreement with JonesTrading Institutional Services LLC, or JonesTrading, pursuant to which the
Company may sell, from time to time, through or to JonesTrading, up to an aggregate of $
On June 28, 2022, the Company entered into an Amendment and Restated Capital on Demand™ Sales Agreement (the “A&R Sales Agreement”) with JonesTrading and B. Riley Securities, Inc. (“B. Riley Securities”). The A&R Sales Agreement modifies the original Capital on Demand™ Sales Agreement to include B. Riley Securities as an additional sales agent thereunder.
2019 Amended and Restated Stock Plan
In December 2019, the Company’s
2019 Stock Plan was established. The expiration date of the plan is October 18, 2029 and the total number of shares of the Company’s
common stock available for grant to employees, directors and consultants of the Company was
2013 Amended and Restated Stock Plan
In September 2013, the Company’s
2013 Stock Plan was established. The expiration date of the plan is September 9, 2023 and at the time of approval, the total number of
shares of the Company’s common stock available for grant to employees, directors and consultants of the Company under the plan was
2013 Equity Incentive Plan
In September 2013, the Company’s
2013 Equity Incentive Plan was established. The expiration date of the plan is September 9, 2023 and the total number of shares of the
Company’s common stock available for grant to employees, directors and consultants of the Company under the plan was
F-13
Stock Options
Following is a summary of stock option activity for the years ended December 31, 2022 and 2021:
| (in thousands, except for per-share amount) | Number of Options | Weighted Average Exercise Price ($) | Weighted Average Remaining Contractual Term (in years) | Aggregate Intrinsic Value ($) | ||||||||||||
| Outstanding, January 1, 2021 | ||||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ( | ) | ||||||||||||||
| Cancelled | ( | ) | ||||||||||||||
| Outstanding, December 31, 2021 | ||||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ||||||||||||||||
| Cancelled | ( | ) | ||||||||||||||
| Outstanding, December 31, 2022 | ||||||||||||||||
| Exercisable, December 31, 2022 | ||||||||||||||||
During 2022, the Company granted
its employees and members of the Board of Directors options to purchase
During 2021, the Company granted
its employees and members of the Board of Directors options to purchase
During the years ended December
31, 2022 and 2021, options to purchase
The fair values of all options
issued and outstanding are being amortized over their respective vesting periods. The unrecognized compensation expense at December 31,
2022 was $
Restricted Stock Units
The Company issued
| (in thousands, except for per-share amount) | RSUs | Weighted Average Grant date Fair Value Per Share ($) | ||||||
| Outstanding, January 1, 2022 | ||||||||
| Granted | ||||||||
| Vested | ||||||||
| Outstanding, December 31, 2022 | ||||||||
The RSUs vest at the earliest
of a change of control event, the termination of the recipient’s continuous service status for any reason other than by the Company
for cause and the third anniversary of the date of the grant. The fair value of the RSUs, $
F-14
Warrants
Following is a summary of warrant activities for the years ended December 31, 2022 and 2021:
| (in thousands, except for per-share amounts) | Number of Warrants | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (in years) | Aggregate Intrinsic Value | ||||||||||||
| Outstanding, January 1, 2021 | ||||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ||||||||||||||||
| Cancelled | ( | ) | ||||||||||||||
| Outstanding, December 31, 2021 | ||||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ||||||||||||||||
| Cancelled | ( | ) | ||||||||||||||
| Outstanding, December 31, 2022 | ||||||||||||||||
| Exercisable, December 31, 2022 | ||||||||||||||||
On August 2, 2022, warrants
to purchase an aggregate of
During the years ended December
31, 2022 and 2021, the Company recorded stock-based compensation expense related to warrants of $
Note 8 - Income Taxes
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets and liabilities at December 31, 2022 and 2021 are as follows:
| (in thousands) | 2022 | 2021 | ||||||
| Deferred tax assets: | ||||||||
| Net operating losses carry forward | $ | $ | ||||||
| Share-based compensation | ||||||||
| Research and development/orphan drug credits | ||||||||
| Capitalized research and development expenses | ||||||||
| Others | ||||||||
| Less: valuation allowance | ( | ) | ( | ) | ||||
| Deferred tax assets, net | $ | $ | ||||||
The Company has recorded a
valuation allowance of $
F-15
For federal income tax purposes,
the Company has $
For state income tax purposes,
the Company has $
The Company has federal research
and development tax credits of $
Federal and state tax laws impose limitations on the utilization of net operating losses and credit carryforwards in the event of an ownership change for tax purposes, as defined in Section 382 of the Internal Revenue Code. Accordingly, the Company’s ability to utilize these carryforwards may be limited as a result of an ownership change which may have already happened or may happen in the future. Such an ownership change could result in a limitation in the use of the net operating losses in future years and possibly a reduction of the net operating losses available.
The difference between the income tax provision and the amount that would result if the U.S. Federal statutory rates were applied to pre-tax losses for the year ended December 31, 2022 and 2021 are as follows:
| (in thousands) | December 31, 2022 | December 31, 2021 | ||||||||||||||
| Federal statutory income taxes | $ | ( | ) | ( | )% | $ | ( | ) | ( | )% | ||||||
| State income taxes | ( | ) | ( | )% | ( | ) | ( | )% | ||||||||
| Deferred true-up | % | % | ||||||||||||||
| Research and development/orphan drug tax credit | ( | ) | ( | )% | ( | ) | ( | )% | ||||||||
| Other | % | % | ||||||||||||||
| Change in valuation allowance | % | % | ||||||||||||||
| Provision for income tax | $ | $ | ||||||||||||||
Note 9 - Subsequent Event
Since December 31, 2022, the
Company has sold
F-16
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE.
None.
ITEM 9A. CONTROLS AND PROCEDURES.
Disclosure controls and procedures. The Company, under the supervision and with the participation of its management, including the Company’s principal executive officer and principal financial and accounting officer, evaluated the effectiveness of the Company’s “disclosure controls and procedures,” as such term is defined in Rule 13a-15(e) and 15d-15(e) under the Securities Act of 1934, as amended (the “Exchange Act”), as of the end of the period covered by this Annual Report on Form 10-K. Based on that evaluation, the Company’s principal executive officer and principal financial and accounting officer have concluded that the Company’s disclosure controls and procedures are effective as of December 31, 2022 to ensure that information required to be disclosed by the Company in reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms, and includes controls and procedures designed to ensure that information required to be disclosed by the Company in such reports is accumulated and communicated to the Company’s management, including the Company’s principal executive officer and principal financial and accounting officer, as appropriate, to allow timely decisions regarding required disclosure.
Management’s Annual Report on Internal Control Over Financial Reporting. The Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting. The Company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
The Company’s internal control over financial reporting includes policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect transactions and dispositions of assets; (2) provide reasonable assurances that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of management and the directors of the Company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on our financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2022. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013). Based on its assessment and those criteria, management concluded that as of December 31, 2022, the Company’s internal control over financial reporting was effective.
This Annual Report on Form 10-K does not include an attestation report from our registered public accounting firm regarding internal control over financial reporting. Our internal control over financial reporting was not subject to such attestation as we are a non-accelerated filer.
Changes in internal controls over financial reporting. There were no changes in the Company’s internal controls over financial reporting that occurred during the fourth quarter of the fiscal year covered by this Annual Report on Form 10-K that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
ITEM 9B. OTHER INFORMATION.
None.
ITEM 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
64
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
Directors and Executive Officers
The names, positions and ages of our directors and executive officers as of March 31, 2023, are as follows:
| Name | Age | Position | ||
| Sandesh Seth | 58 | Chairman and Chief Executive Officer | ||
| Steve O’Loughlin | 38 | Chief Financial Officer (Principal Financial and Accounting Officer) | ||
| Jeffrey W. Chell M.D. | 68 | Director | ||
| David Nicholson, Ph.D. | 67 | Lead Independent Director | ||
| Richard I. Steinhart | 65 | Director | ||
| Ajit S. Shetty, Ph.D. | 76 | Director |
Directors hold office for a term consistent with classified board provisions of our Charter. For further information, see the section titled “—Corporate Governance—Term of Office” below. Officers serve at the discretion of the Board of Directors.
There are no other arrangements or understanding between any of our directors and any other persons pursuant to which they were selected as a director.
Background of Executive Officers and Directors
The principal occupations for the past five years (and, in some instances, for prior years) of each of our directors and executive officers are as follows:
Sandesh Seth, Chairman and Chief Executive Officer
Mr. Sandesh Seth has been our Chief Executive Officer since June 2017. Mr. Seth has been a Director since March 2012, our Chairman of the Board since October 2013, and served as Executive Chairman from August 2014 to June 2017.
Mr. Seth has 25+ years of experience in investment banking (Laidlaw& Co (UK) Ltd., Cowen & Co.), equity research (Bear Stearns, Commonwealth Associates) and in the pharma industry (Pfizer, Warner-Lambert, SmithKline in strategic planning, business development and R&D project management). Mr. Seth was chairman of Relmada Therapeutics Inc., a specialty pharma company focused on CNS therapeutics, which he helped co-found. Mr. Seth has an MBA in Finance from New York University; an M.S. in the Pharmaceutical Sciences from the University of Oklahoma Health Center and a B.Sc. in Chemistry from Bombay University. He has published several scientific articles and was awarded the University Regents Award for Research Excellence at the University of Oklahoma. Mr. Seth was designated as Regulatory Affairs Certified by the Regulatory Affairs Professionals Society which signifies proficiency with U.S. FDA regulations. He has several patents related to use of radiopharmaceuticals as conditioning agents for adoptive cell therapies and as therapeutic combinations.
That Mr. Seth has served in various business executive-level positions over the course of his career, has significant investment banking experience, has developed significant management, operational and leadership skills and is well accustomed to interfacing with investors, analysts, auditors, C-level executives, and outside advisors, led us to conclude that Mr. Seth should serve as a director.
65
Steve O’Loughlin, Chief Financial Officer
Steve O’Loughlin has been our Chief Financial Officer since August 2020. Mr. O’Loughlin served as our Principal Financial Officer from May 2017 to August 2020. Mr. O’Loughlin joined Actinium in October 2015 as Vice President, Finance and Corporate Development, with almost a decade of life sciences industry experience gained from previous positions in investment banking and publicly traded life sciences companies. Prior to Actinium, from June 2015 to October 2015, Mr. O’Loughlin worked at J. Streicher LLC as an investment banker, from August 2012 to June 2015 Mr. O’Loughlin held the position of vice president, corporate finance and development and was a corporate officer at Protea Biosciences, Inc., a publicly traded life sciences tools company. Previously, From June 2010 to June 2012, Mr. O’Loughlin held corporate development positions with Caliber I.D., a publicly traded diagnostics company. Mr. O’Loughlin previously worked in investment banking at Jesup & Lamont where he focused on the biotechnology and life sciences industries. Mr. O’Loughlin has a B.S. in Business Administration with a concentration in finance from Ramapo College of New Jersey.
Jeffrey W. Chell, M.D., Director
Dr. Chell has been a Director of the Company since April 2018. Dr. Chell is also a member of our Audit Committee and Compensation Committee. He has been the chief executive officer emeritus of the National Marrow Donor Program (“NMDP”) since 2017 having served as its chief executive officer since 2000. Dr. Chell has led the NMDP through transformational growth as its Be The Match Registry tripled to more than 12 million donors, the number of transplants facilitated has grown fivefold to over 6,400 annually, and revenue more than tripled to nearly $400 million per year. He is also the co-founder and has served as executive director of the Center For International Blood & Marrow Transplant Research since 2004, a leading research program in the field contributing over 70 research publications per year in peer-reviewed journals. Dr. Chell also currently serves as chair of CLR Insurance, a captive insurance company domiciled in the Cayman Islands. From 2014 to 2016, Dr. Chell served as co-chair of Bone Marrow Donors Worldwide during its IT transformation project, improving revenues and reducing costs.
Prior to joining the NMDP, he served as president, Allina Medical Clinics, a 450 physician multi-specialty medical group from 1994 to 1999. Prior to that he practiced Internal Medicine in Minneapolis and in the U.S. Air Force Medical Corps.
Dr. Chell received his M.D. from the University of Minnesota and his training in Internal Medicine at the University of Wisconsin, Madison. Dr. Chell is a diplomate of the American Board of Internal Medicine, a member of the American Society of Hematology and a member of the American Society of Blood and Marrow Transplantation.
He has received multiple honors including the 2018 Public Service award of the American Society For Blood and Marrow Transplantation, 2017 Most Admired CEO by the Minneapolis/St. Paul Business Journal, 2010 Healthcare Executive of the Year by the Minneapolis/St, Paul Business Journal, and the 2017 Bone Marrow Foundation Service Award.
That Dr. Chell brings many years of experience with patient donor programs, knowledge of challenges related to bone marrow transplants, leadership of organizations and experience working in medical groups to our Board, led us to conclude that Dr. Chell should serve as a director.
David Nicholson, Ph.D., Director
David Nicholson has been a Director of the Company since 2008. Dr. Nicholson is also a member of our Compensation Committee and our Nominating and Corporate Governance Committee. Since March 2015, Dr. Nicholson served as Executive Vice President and Chief R&D Officer of Allergan, which was acquired by Abbvie in May 2020. In August 2014, Dr. Nicholson joined Allergan (previously known as Actavis plc and Forest Laboratories, Inc.) as senior vice president, Actavis Global Brands R&D. From March 2012 to August 2014, Dr. Nicholson was on the executive committee of Bayer CropScience as head of research & development responsible for the integration of the company’s R&D activities into one global organization. Dr. Nicholson graduated in pharmacology, earning his B.Sc. from the University of Manchester (1975) and his Ph.D. from the University of Wales (1980). Between 1978 and 1988, Dr. Nicholson worked in the pharmaceutical industry for the British company Beecham-Wülfing in Gronau, Germany. The main emphasis of his activities as group leader in a multidisciplinary project group was the development of cardiovascular drugs.
66
From 1988-2007, Dr. Nicholson held various positions of increasing seniority in the UK, the Netherlands and the U.S. with Organon, a business unit of Akzo Nobel. Ultimately, he became executive vice president, research & development, and member of the Organon Executive Management Committee. He implemented change programs, leading to maximizing effectiveness in research & development, ensuring customer focus and the establishment of a competitive pipeline of innovative drugs. In 2007, Dr. Nicholson transferred to Schering-Plough, Kenilworth, New Jersey as senior vice president, responsible for Global Project Management and Drug Safety. From 2009 to December 2011, he was vice president licensing and knowledge management at Merck in Rahway, New Jersey, reporting to the president of Merck R&D. As an integration team member, Dr. Nicholson played a role in the strategic mergers of Organon BioSciences, the human and animal health business of Dutch chemical giant Akzo-Nobel, and Schering-Plough in 2007 as well as of Schering-Plough and Merck in 2009.
That Dr. Nicholson brings over 25 years of pharmaceutical experience to our Board, having served in various pharmaceutical research and development executive-level positions over the course of his career, and that Dr. Nicholson has developed significant management and leadership skills relating to the pharmaceutical industry and is well accustomed to interfacing with investors, analysts, auditors, outside advisors and governmental officials, led us to conclude that Dr. Nicholson should serve as a director.
Ajit S. Shetty, Ph.D., Director
Dr. Shetty has been a Director of the Company since March 2017. Dr. Shetty is also a member of our Audit Committee, Compensation Committee, and Chairman of our Nominating and Corporate Governance Committee. Dr. Shetty joined Janssen Pharmaceutical, Inc. (“Janssen”) in 1976 ultimately rising to the position of president in 1986 where he led the establishment of Janssen’s business in the U.S. From 1999 to 2008 he was managing director of Janssen, during this time the Janssen Group of companies’ global sales grew from $1 billion to $8 billion, and from 2004 until 2012 he was chairman of the board of directors. In Dr. Shetty’s most recent role at Johnson & Johnson he was head of Enterprise Supply Chain, where he reported to the chief executive officer and was responsible for the transformation and optimization of Johnson & Johnson’s supply chain. Dr. Shetty earned a Ph.D. in Metallurgy and B.A. Natural Sciences from Trinity College, Cambridge University and a Master of Business Administration from Carnegie Mellon University. Dr. Shetty has served as a member of Agile Therapeutics, Inc.’s board of directors since February 2016. In 2007, Dr. Shetty was bestowed the title of Baron by King Albert II of Belgium for his exceptional merits. He is a member of the Board of Trustees of Carnegie Mellon University, serves on the Board of Governors for GS1 (Global Standards) in Belgium and formerly served on the Corporate Advisory Board of the John Hopkins Carey Business School. In 2016, Dr. Shetty was named as chairperson of the Vlaams Instituut voor Biotechnologie (VIB), a Belgium based life sciences research institute focused on translating scientific results into pharmaceutical, agricultural and industrial applications. In addition, he was elected Manager of the Year in 2004 in Flanders and received a Life-Time Achievement Award in India in 2010. We believe Dr. Shetty’s qualifications to sit on our Board include his extensive pharmaceutical experience leading commercial and supply chain operations and his significant education background.
That Dr. Shetty has more than 30 years of leadership and executive experience in the pharmaceutical industry, that he has significant supply chain knowledge and that he has experience conducting business in the U.S. and Europe, led us to conclude that Dr. Shetty should serve as a director.
Richard I. Steinhart, Director
Mr. Steinhart has served as our Director and Chairman of the Audit Committee since November 2013. Mr. Steinhart is also a member of our Nominating and Corporate Governance Committee. Since October 2017 Mr. Steinhart has been the senior vice president and chief financial officer of BioXcel Therapeutics, Inc. Since March 2014, Mr. Steinhart has been a member of the board of directors of Atossa Genetics, Inc. where he is chairman of the audit committee and a member of the compensation committee. From October 2015 to April 2017, Mr. Steinhart was vice president and chief financial officer at Remedy Pharmaceuticals, a privately-held, clinical stage pharmaceutical company. From January 2014 through September 2015 Mr. Steinhart worked as a financial and strategic consultant to the biotechnology and medical device industries. From April 2006 through December 2013, Mr. Steinhart was employed by MELA Sciences, Inc., as its vice president, finance and chief financial officer, treasurer and secretary. In April 2012, Mr. Steinhart received a promotion to senior vice president, finance and chief financial officer. From May 1992 until joining MELA Sciences, Mr. Steinhart was a managing director of Forest Street Capital/SAE Ventures, a boutique investment banking, venture capital, and management consulting firm focused on healthcare and technology companies. Prior to Forest Street Capital/SAE Ventures, he was vice president and chief financial officer of Emisphere Technologies, Inc. Mr. Steinhart’s other experience includes seven years at CW Group, Inc., a venture capital firm focused on medical technology and biopharmaceutical companies, where he was a general partner and chief financial officer. Mr. Steinhart began his career at Price Waterhouse, now known as PricewaterhouseCoopers. He holds BBA and MBA degrees from Pace University and is a Certified Public Accountant (inactive).
That Mr. Steinhart brings more than 30 years of financial experience to our Board, having served in various executive-level financial positions over the course of his career, and that Mr. Steinhart is a certified public accountant (inactive), led us to conclude that Mr. Steinhart should serve as a director and chair the Audit Committee.
67
Corporate Governance
Our Board of Directors oversees our business affairs and monitors the performance of management. In accordance with our corporate governance principles, our Board of Directors does not involve itself in day-to-day operations. The Directors keep themselves informed through discussions with the Chairman and Chief Executive Officer and other key executives and by reading the reports and other materials that we send them and by participating in Board of Directors and committee meetings.
Term of Office
Our directors are divided into three classes, designated Class I, Class II and Class III. Class I shall consists of two directors, Class II shall consist of one director, and Class III consists of one director. The term of office for each Class I director expires at 2023 Annual Meeting of Stockholders; the term of office for each Class II director expires at the 2024 Annual Meeting of stockholders; and the term of office for each Class III director expires at the 2025 Annual Meeting of stockholders.
The term of each director is set forth below or until their successors are duly elected:
| Director | Class | Term (from 2022 Annual Meeting) | ||
| David Nicholson | Class I | 1 year | ||
| Richard Steinhart | Class I | 1 year | ||
| Sandesh Seth | Class II | 2 years | ||
| Jeffrey W. Chell | Class II | 2 years | ||
| Ajit Shetty | Class III | 3 years |
Notwithstanding the foregoing, each director shall serve until his successor is duly elected and qualified, or until his retirement, death, resignation or removal.
Director Independence
We use the definition of “independence” of the NYSE American stock exchange to make this determination. We are listed on the NYSE American under the symbol “ATNM”. NYSE MKT corporate governance rule Sec. 803(A)(2) provides that an “independent director” means a person other than an executive officer or employee of the company. No director qualifies as independent unless the issuer’s board of directors affirmatively determines that the director does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. Under the NYSE American director independence rules, Jeffrey W. Chell, David Nicholson, Ajit S. Shetty, and Richard I. Steinhart are independent directors of the Company.
68
Chief Executive Officer Compensation
On August 12, 2020, we and Mr. Seth entered into an employment agreement whereby Mr. Seth will serve as Chairman and Chief Executive Officer until February 24, 2024, unless terminated earlier as set forth in the employment agreement.
Under the terms of the employment agreement, Mr. Seth is entitled to (i) a base salary, which will be determined by the Board and adjusted to be competitively aligned to a range between the 25th and 75th percentile of the relevant market data of chief executive officer positions of similarly situated publicly companies, (ii) a performance bonus with a target of 50% of his annual base salary as well as other multipliers as determined by the Board and (iii) options to purchase shares of common stock of the Company as the Board may grant. For 2021, Mr. Seth’s annual base salary was set at $615,000, and for 2022, his annual base salary was set at $665,000.
When and if granted, options will have an exercise price equal to the closing price of the Company’s common stock on the date of the approval, and 2% of the grant will vest each month from the grant date until fully vested, in accordance with the 2013 Stock Plan and 2019 Plan. The options will expire 10 years from the grant date, subject to Mr. Seth’s continuing service with the Company. Mr. Seth also receives the standard benefits available to other similarly situated employees.
If Mr. Seth’s employment as Chief Executive Officer or Chairman is terminated due to death or disability, Mr. Seth will be entitled to earned, but unpaid, salary, benefits and the Pro-Rated Bonus (as defined below) for the year of termination. Upon termination of his employment for Cause (as defined in the employment agreement), or his resignation without Good Reason (as defined in the employment agreement), Mr. Seth will receive any accrued and unpaid base salary, the Pro-Rated Bonus and benefits through the date of termination.
If we terminate Mr. Seth’s employment without Cause, or if Mr. Seth resigns for Good Reason other than in connection with a Change in Control, Mr. Seth will be entitled to (i) a single lump sum payment equal to 24 months of his compensation, (ii) continued health benefits for 24 months, (iii) immediate vesting of all outstanding equity awards granted to Mr. Seth, and (iv) a single lump sum payment equal to his annual bonus subject to the achievement of the applicable goals, pro-rated based on the number of days in the Company’s fiscal year through the date of termination (the “Pro-Rated Bonus”).
In addition, if we terminate Mr. Seth’s employment without Cause or if Mr. Seth resigns for Good Reason, or if we fail to renew his position as Chief Executive Officer and Chairman on February 24, 2024, in any case, within the 12-month period beginning on the date of a Change in Control (as defined in the 2013 Stock Plan and 2019 Plan), Mr. Seth will be entitled to (i) a single lump sum payment equal to 30 months of his compensation, (ii) continued health benefits for 30 months, (iii) immediate vesting of all outstanding equity awards granted to Mr. Seth, and (iv) a single lump sum payment equal to the Pro-Rated Bonus.
Chief Financial Officer Compensation
On August 12, 2020, we entered into an employment agreement with Mr. O’Loughlin, pursuant to which he serves as Chief Financial Officer of the Company. Under the terms of the employment agreement, Mr. O’Loughlin is entitled to (i) a base salary, which shall be determined by the Board, (ii) a performance bonus, which may be up to 30% of the annual base salary based upon the achievement of certain objectives such as the Board shall determine and (iii) options to purchase shares of common stock of the Company as the Board may grant. For 2021, Mr. O’Loughlin’s annual base salary was set at $370,000, and for 2022, his annual base salary was set at $400,000.
When and if granted, options will have an exercise price equal to the closing price of the Company’s common stock on the date of the approval, and 2% of the grant will vest each month from the grant date until fully vested, in accordance with the 2013 Stock Plan and 2019 Plan. The options will expire 10 years from the grant date, subject to Mr. O’Loughlin’s continuing service with the Company. Mr. Loughlin will also receive the standard benefits available to other similarly situated employees.
In addition, if we terminate Mr. O’Loughlin’s employment without Cause (as defined in the employment agreement) or if Mr. O’Loughlin resigns for Good Reason (as defined in the employment agreement), in either case, within the 12-month period beginning on the date of a Change in Control, Mr. O’Loughlin will be entitled to (i) a single lump sum payment equal to his annual base salary, (ii) continued health benefits for 12 months, and (iii) immediate vesting of all outstanding equity awards granted to Mr. O’Loughlin.
69
Board of Directors Meetings and Attendance
During 2022, our Board of Directors held five meetings and acted by unanimous written consent on three occasions. Each director attended all of the meetings of our Board during the year ended December 31, 2022.
Committees of the Board of Directors
Our Board of Directors has formed three standing committees: Audit, Compensation and Nominating and Corporate Governance. Actions taken by our committees are reported to the full board. Each of our committees has a charter and each charter is posted on our website.
| Audit Committee | Compensation Committee | Nominating and Corporate Governance Committee | ||
| Richard I. Steinhart* | David Nicholson* | Ajit S. Shetty* | ||
| Jeffrey W. Chell | Jeffrey W. Chell | David Nicholson | ||
| Ajit S. Shetty | Ajit S. Shetty | Richard I. Steinhart |
| * | Indicates committee chair |
Audit Committee
Our Audit Committee, which currently consists of three independent directors, provides assistance to our Board in fulfilling its legal and fiduciary obligations with respect to matters involving the accounting, financial reporting, internal control and compliance functions of the Company. The Board has determined that Mr. Steinhart is an “audit committee financial expert” as defined in Item 407(d)(5)(ii) of Regulation S-K. Our Audit Committee employs an independent registered public accounting firm to audit the financial statements of the Company and perform other assigned duties. Further, our Audit Committee provides general oversight with respect to the accounting principles employed in financial reporting and the adequacy of our internal controls. In discharging its responsibilities, our Audit Committee may rely on the reports, findings and representations of the Company’s auditors, legal counsel, and responsible officers. Our Board has determined that all members of the Audit Committee are financially literate within the meaning of SEC rules and under the current listing standards of the NYSE American. The Audit Committee met four times during 2022. Each member of the Audit Committee was present at all of the Audit Committee meetings held during 2022.
Compensation Committee
Our Compensation Committee, which currently consists of three directors, establishes executive compensation policies consistent with the Company’s objectives and stockholder interests. The Compensation Committee met two times and acted by unanimous written consent on one occasion during 2022. Each member of the Compensation Committee was present at all committee meetings held in 2022. Our Compensation Committee also reviews the performance of our executive officers and establishes, adjusts and awards compensation, including incentive-based compensation, as more fully discussed below. In addition, our Compensation Committee generally is responsible for:
| ● | establishing and periodically reviewing our compensation philosophy and the adequacy of compensation plans and programs for our directors, executive officers and other employees; |
| ● | overseeing our compensation plans, including the establishment of performance goals under the Company’s incentive compensation arrangements and the review of performance against those goals in determining incentive award payouts; |
| ● | overseeing our executive employment contracts, special retirement benefits, severance, change in control arrangements and/or similar plans; |
| ● | acting as administrator of any company stock option plans; and |
| ● | overseeing outside compensation consultants when engaged. |
70
Our Compensation Committee periodically reviews the compensation paid to our non-employee directors and the principles upon which their compensation is determined. The Compensation Committee also periodically reports to the Board on how our non-employee director compensation practices compare with those of other similarly situated public corporations and, if the Compensation Committee deems it appropriate, recommends changes to our director compensation practices to our Board for approval.
Outside consulting firms retained by our Compensation Committee and management also will, if requested, provide assistance to the Compensation Committee in making its compensation-related decisions. The Compensation Committee engaged StreeterWyatt Analytics LLC, or Streeter Wyatt and paid consultant fees of $22,000 during the year ended December 31, 2022. Streeter Wyatt was instructed to provide support and analysis to the Compensation Committee and their services included developing a peer group regarding executive and director compensation.
Nominating and Corporate Governance Committee
Our Nominating and Corporate Governance Committee, which currently consists of three directors, is charged with the responsibility of reviewing our corporate governance policies and proposing potential director nominees to the Board for consideration. Our Board has determined that each member of our Nominating and Corporate Governance Committee qualifies as an “independent” member of the Board as defined by the rules and regulations of the SEC and the NYSE American. The Nominating and Corporate Governance Committee held no meetings and acted by unanimous written consent on one occasion during 2022.
Our Nominating and Corporate Governance Committee’s primary responsibilities and obligations include, among other things:
| ● | overseeing the administration of our Code of Business Ethics and Conduct and related policies; |
| ● | leading the search for and recommending individuals qualified to become members of the Board, and selecting director nominees to be presented for election by the shareholders at each annual meeting; |
| ● | ensuring, in cooperation with the Compensation Committee, that no agreements or arrangements are made with directors or relatives of directors for providing professional or consulting services to us or our affiliate or individual officer or one of their affiliated, without appropriate review and evaluation for conflicts of interest; |
| ● | assessing the independence of directors annually and report to the Board; |
| ● | recommending to the Board for its approval, the leadership structure of the Board, including whether the Board should have an executive or non-executive Chairman, whether the roles of Chairman and Chief Executive Officer should be combined, and whether a Lead Director of the Board should be appointed; provided that such structure shall be subject to the bylaws of the Company then in effect; |
| ● | ensuring that Board members do not serve on more than six other for-profit public company boards that have a class of securities registered under the Exchange Act in addition to the Board; |
| ● | reviewing the Board’s committee structure and to recommend to the Board for its approval directors to serve as members of each committee as well as recommendations for committee chairs; |
| ● | reviewing and recommending changes to procedures whereby shareholders may communicate with the Board; |
| ● | reviewing recommendations received from shareholders for persons to be considered for nomination to the Board; |
| ● | monitoring compliance with our corporate governance guidelines; |
| ● | developing and implementing an annual self-evaluation of the Board, both individually and as a Board, and of its committees; |
71
Our Nominating and Corporate Governance Committee considers all qualified candidates identified by members of the Board, by senior management and by stockholders. The Committee follows the same process and uses the same criteria for evaluating candidates proposed by stockholders, members of the Board and members of senior management. When evaluating a candidate to serve on our Board, the members of our Nominating and Corporate Governance Committee consider items such as experience in the biotechnology sector, experience with public companies, executive managerial experience, operations and commercial experience, fundraising experience and contacts in the investment banking industry, personal and skill set compatibility with current Board members, industry reputation, knowledge of our company generally, and independence.
Our Amended and Restated Bylaws, as amended (the “Bylaws”) contains provisions that address the process by which a stockholder may nominate an individual to stand for election to the Board at our annual meetings. To recommend a nominee for election to the Board, a stockholder must submit his or her recommendation to our Secretary at our corporate offices at 275 Madison Avenue, 7th Floor, New York, New York 10016. Such nomination must satisfy the notice, information and consent requirements set forth in our Bylaws and must be received by us prior to the date set forth under “Submission of Future Stockholder Proposals” below. A stockholder’s recommendation must be accompanied by the information with respect to stockholder nominees as specified in our Bylaws, including among other things, the name, age, address and occupation of the recommended person, the proposing stockholder’s name and address, the ownership interests of the proposing stockholder and any beneficial owner on whose behalf the nomination is being made (including the number of shares beneficially owned, any hedging, derivative, short or other economic interests and any rights to vote any shares) and any material monetary or other relationships between the recommended person and the proposing stockholder and/or the beneficial owners, if any, on whose behalf the nomination is being made.
Our approach toward Board diversity takes into consideration the overall composition and diversity of the Board and areas of expertise that director nominees may be able to offer, including business experience, knowledge, abilities, customer relationships and appropriate perspectives on environmental, social and governance matters. Generally, we strive to assemble and maintain a Board that brings to us a variety of perspectives and skills derived from business and professional experience as we may deem are in our and our stockholders’ best interests. In doing so, we also consider candidates with appropriate non-business backgrounds.
Lead Director
In September 2017, our Board of Directors created the position of Lead Director and designated David Nicholson, an existing independent director, as our Lead Director. Pursuant to the charter of the Lead Director, the Lead Director shall be an independent, non-employee director designated by our Board of Directors who shall serve in a lead capacity to coordinate the activities of the other non-employee directors, interface with and advise management, and perform such other duties as are specified in the charter or as our Board of Directors may determine.
Family Relationships
There are no family relationships among any of our officers or directors.
Involvement in Certain Legal Proceedings
To our knowledge, none of our current directors or executive officers has, during the past ten years:
| ● | been convicted in a criminal proceeding or been subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); |
| ● | had any bankruptcy petition filed by or against the business or property of the person, or of any partnership, corporation or business association of which he was a general partner or executive officer, either at the time of the bankruptcy filing or within two years prior to that time; |
| ● | been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction or federal or state authority, permanently or temporarily enjoining, barring, suspending or otherwise limiting, his involvement in any type of business, securities, futures, commodities, investment, banking, savings and loan, or insurance activities, or to be associated with persons engaged in any such activity; |
72
| ● | been found by a court of competent jurisdiction in a civil action or by the SEC or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; |
| ● | been the subject of, or a party to, any federal or state judicial or administrative order, judgment, decree, or finding, not subsequently reversed, suspended or vacated (not including any settlement of a civil proceeding among private litigants), relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies including, but not limited to, a temporary or permanent injunction, order of disgorgement or restitution, civil money penalty or temporary or permanent cease-and-desist order, or removal or prohibition order, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or |
| ● | been the subject of, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization (as defined in Section 3(a)(26) of the Exchange Act), any registered entity (as defined in Section 1(a)(29) of the Commodity Exchange Act), or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
None of our directors or executive officers has been involved in any transactions with us or any of our directors, executive officers, affiliates or associates which are required to be disclosed pursuant to the rules and regulations of the SEC.
Code of Ethics
The Company has adopted a code of ethics, a copy of which is attached as Exhibit 14.1 to the Form 8-K filed on January 2, 2013.
73
Compensation Discussion and Analysis
Our Compensation Committee of our Board of Directors has the responsibility to review, determine and approve the compensation for our executive officers. Further, our Compensation Committee oversees our overall compensation strategy, including compensation policies, plans and programs that cover all employees. At our 2022 Annual Meeting of Stockholders, our Stockholders voted on an advisory basis to approve the compensation of named executive officers. Of the votes cast (excluding abstentions and broker non-votes), 79.3% were cast in support of the results of our compensation program. In light of this, in reviewing the executive compensation program for 2021 and 2022, our Compensation Committee decided to retain the general overall program design, which ties a significant portion of the executives’ pay closely with our performance. In the future, our Compensation Committee will continue to consider the executive compensation program in light of changing circumstances and stockholder feedback.
We currently employ two executive officers: (1) Sandesh Seth, our Chairman and Chief Executive Officer (who we refer to in this Compensation Discussion and Analysis as our CEO) and (2) Steve O’Loughlin, our Chief Financial Officer.
This Compensation Discussion and Analysis sets forth a discussion of the compensation for our Named Executive Officers, or NEOs, as well as a discussion of our philosophies underlying the compensation for our NEOs and our employees generally.
Objectives of Our Compensation Program
The Compensation Committee’s philosophy seeks to align the interests of our stockholders, officers and employees by tying compensation to individual and company performance, both directly in the form of salary or annual cash incentive payments, and indirectly in the form of equity awards. The objectives of our compensation program enhance our ability to:
| ● | attract and retain qualified and talented individuals; and |
| ● | provide reasonable and appropriate incentives and rewards to our team for building long-term value within our company, in each case in a manner comparable to companies similar to ours. |
In addition, we strive to be competitive with other similarly situated companies in our industry. The process of developing pharmaceutical products and bringing those products to market is a long-term proposition and outcomes may not be measurable for several years. Therefore, in order to build long-term value for our company and its stockholders, and in order to achieve our business objectives, we believe that we must compensate our officers and employees in a competitive and fair manner that reflects current company activities but also reflects contributions to building long-term value.
We utilize the services of StreeterWyatt Analytics LLC to review compensation programs of peer companies in order to assist the Compensation Committee in determining the compensation levels for our NEOs, as well as for other employees of our company. StreeterWyatt is a recognized independent consulting company and services clients throughout the United States.
Elements of Our Compensation Program and Why We Chose Each
Main Compensation Components
Our company-wide compensation program, including for our NEOs, is broken down into three main components: base salary, performance cash bonuses and potential long-term compensation in the form of stock options or restricted stock unit awards. We believe these three components constitute the minimum essential elements of a competitive compensation package in our industry.
Salary
Base salary is used to recognize the experience, skills, knowledge and responsibilities required of our NEOs as well as recognizing the competitive nature of the biopharmaceutical industry. This is determined partially by evaluating our peer companies as well as the degree of responsibility and experience levels of our NEOs and their overall contributions to our company. Base salary is one component of the compensation package for NEOs; the other components being cash bonuses, annual equity grants, and company benefit programs. Base salary is determined in advance whereas the other components of compensation are awarded in varying degrees following an assessment of the performance of a NEO. This approach to compensation reflects the philosophy of our Board of Directors and its Compensation Committee to emphasize and reward, on an annual basis, performance levels achieved by our NEOs.
74
Performance Bonus Plan
We have a performance bonus plan under which bonuses are paid to our NEOs based on achievement of company performance goals and objectives established by the Compensation Committee and/or our Board of Directors as well as on individual performance. The bonus program is discretionary and is intended to: (i) strengthen the connection between individual compensation and our company’s achievements; (ii) encourage teamwork among all disciplines within our company; (iii) reinforce our pay-for-performance philosophy by awarding higher bonuses to higher performing employees; and (iv) help ensure that our cash compensation is competitive. Depending on the cash position of the company, the Compensation Committee and our Board of Directors have the discretion to not pay cash bonuses in order that we may conserve cash and support ongoing development programs and commercialization efforts. Regardless of our cash position, we consistently grant annual merit-based stock options to continue incentivizing both our senior management and our employees.
Based on their employment agreements, each NEO is assigned a target payout under the performance bonus plan, expressed as a percentage of base salary for the year. Actual payouts under the performance bonus plan are based on the achievement of corporate performance goals and an assessment of individual performance, each of which is separately weighted as a component of such officer’s target payout. For the NEOs, the corporate goals receive the highest weighting in order to ensure that the bonus system for our management team is closely tied to our corporate performance. Each employee also has specific individual goals and objectives as well that are tied to the overall corporate goals. For employees, mid-year and end-of-year progress is reviewed with the employees’ managers.
Equity Incentive Compensation
We view long-term compensation, currently in the form of stock options generally vesting in annual increments over four years, as a tool to align the interests of our NEOs and employees generally with the creation of stockholder value, to motivate our employees to achieve and exceed corporate and individual objectives and to encourage them to remain employed by the company. While cash compensation is a significant component of employees’ overall compensation, the Compensation Committee and our Board of Directors (as well as our NEOs) believe that the driving force of any employee working in a small biotechnology company should be strong equity participation. We believe that this not only creates the potential for substantial longer-term corporate value but also serves to motivate employees and retain their loyalty and commitment with appropriate personal compensation.
Other Compensation
In addition to the main components of compensation outlined above, we also have provided contractual severance and/or change in control benefits to several employees including our CEO. The change in control benefits for all applicable persons have a “double trigger.” A double-trigger means that the executive officers will receive the change in control benefits described in the agreements only if there is both (1) a Change in Control of our company (as defined in the agreements) and (2) a termination by us of the applicable person’s employment “without cause” or a resignation by the applicable persons for “good reason” (as defined in the agreements) within a specified time period prior to or following the Change in Control. We believe this double trigger requirement creates the potential to maximize stockholder value because it prevents an unintended windfall to management as no benefits are triggered solely in the event of a Change in Control while providing appropriate incentives to act in furtherance of a change in control that may be in the best interests of the stockholders. We believe these severances or change in control benefits are important elements of our compensation program that assist us in retaining talented individuals at the executive and senior managerial levels and that these arrangements help to promote stability and continuity of our executives and senior management team. Further, we believe that the interests of our stockholders will be best served if the interests of these members of our management are aligned with theirs. We believe that providing change in control benefits lessens or eliminates any potential reluctance of members of our management to pursue potential change in control transactions that may be in the best interests of the stockholders. We also believe that it is important to provide severance benefits to members of our management, to promote stability and focus on the job at hand.
We also provide benefits to the executive officers that are generally available to all regular full-time employees of our company, including our medical and dental insurance, and a 401(k) plan. Further, we do not have deferred compensation plans, pension arrangements or post-retirement health coverage for our executive officers or employees. All of our employees not specifically under contract are “at-will” employees, which means that their employment can be terminated at any time for any reason by either us or the employee.
75
Determination of Compensation Amounts
A number of factors impact the determination of compensation amounts for our NEOs, including the individual’s role in the company and individual performance, length of service with the company, competition for talent, individual compensation package, assessments of internal pay equity and industry data. Stock price performance has generally not been a factor in determining annual compensation because the price of our common stock is subject to a variety of factors outside of our control.
Industry Survey Data
In collaboration with StreeterWyatt, we establish and maintain a list of peer companies to best assure ourselves that we are compensating our executives on a fair and reasonable basis, as set forth above under the heading “Objectives of our Compensation Program.” We also utilize StreeterWyatt-prepared data for below-executive level personnel, which data focuses on biotechnology companies that can be considered peers in terms of numerous variables including phase of development, size, therapeutic and technological focus among others. The availability of peer data is used by the Compensation Committee strictly as a guide in determining compensation levels with regard to salaries, cash bonuses and performance related annual equity grants to all employees. However, the availability of this data does not imply that the Compensation Committee is under any obligation to exactly follow peer companies in compensation matters.
Determination of Base Salaries
As a guideline for NEO base salary, we perform formal benchmarks against respective comparable positions in our established peer group. We adjust salaries based on our assessment of our NEOs’ levels of responsibility, experience, overall compensation structure and individual performance. The Compensation Committee is not obliged to raise salaries purely on the availability of data. Merit-based increases to salaries of executive officers are based on our assessment of individual performance and the relationship to applicable salary ranges. Cost of living adjustments may also be a part of that assessment.
Performance Bonus Plan
Concurrently with the beginning of each calendar year, preliminary corporate goals that reflect our business priorities for the coming year are prepared by the CEO with input from the other executive officers. These goals are weighted by relative importance. The draft goals and proposed weightings are presented to the Compensation Committee and the Board and discussed, revised as necessary, and then approved by our Board of Directors. The Compensation Committee then reviews the final goals and their weightings to determine and confirm their appropriateness for use as performance measurements for purposes of the bonus program. The goals and/or weightings may be re-visited during the year and potentially restated in the event of significant changes in corporate strategy or the occurrence of significant corporate events. Following the agreement of our Board on the corporate objectives, the goals are then shared with all employees in formal meetings and are reviewed periodically throughout the year.
Determination of Equity Incentive Compensation
To assist us in assessing the reasonableness of our equity grant amounts, we have reviewed StreeterWyatt supplied information. Such information included equity data from a cross-section of similar companies in our industry.
76
Equity Grant Practices
All stock options and/or restricted stock units granted to the NEOs and other executives are approved by the Compensation Committee. Exercise prices for options are set at the closing price of our common stock on the date of grant. Grants are generally made: (i) on the employee’s start date and (ii) at board of director meetings held once each year and following annual performance reviews. However, grants have been made at other times during the year. The size of year-end grants for each NEO is assessed against our internal equity guidelines. Current market conditions for grants for comparable positions and internal equity may also be assessed. Also, grants may be made in connection with promotions or job-related changes in responsibilities. In addition, on occasion, the Compensation Committee may make additional special awards for extraordinary individual or company performance.
Compensation Setting Process
Annually, at a meeting of our Board of Directors and the Compensation Committee, overall corporate performance and relative achievement of the corporate goals for the prior year are assessed. The relative achievement of each goal is assessed and quantified and the summation of the individual components results in a corporate goal rating, expressed as percentages. The Compensation Committee then approves the final disbursement of salary increases, cash bonuses and option or restricted stock unit grants.
The Compensation Committee looks to the CEO’s performance assessments of the other NEOs and his recommendations regarding a performance rating for each, as well as input from the other members of our Board of Directors. These recommendations may be adjusted by the Compensation Committee prior to finalization. For the CEO, the Compensation Committee evaluates his performance, taking into consideration input from the other members of our Board of Directors, and considers the achievement of overall corporate objectives by both the CEO specifically and the company generally. The CEO is not present during the Compensation Committee’s deliberations regarding his compensation.
The Compensation Committee has the authority to directly engage, at our company’s expense, any compensation consultants or other advisors (such as StreeterWyatt) that it deems necessary to determine the amount and form of employee, executive and director compensation. In determining the amount and form of employee, executive and director compensation, the Compensation Committee has reviewed and discussed historical salary information as well as salaries for similar positions at comparable companies. However, the availability of this data does not imply that the Compensation Committee is under any obligation to exactly follow peer companies’ compensation practices.
We paid consultant fees to StreeterWyatt of $22,000 during the year ended December 31, 2022. NEOs may have indirect input in the compensation results for other executive officers by virtue of their participation in the performance review and feedback process for the other executive officers.
77
ITEM 11. EXECUTIVE COMPENSATION
Summary Compensation Table
The following table provides information regarding the compensation earned during the years ended December 31, 2022 and 2021 for our named executive officers.
| Name/Position | Year | Salary | Bonus (1) | Option Awards (2) |
All Other Compensation |
Total | ||||||||||||||||
| Sandesh Seth | 2022 | $ | 665,000 | $ | 480,000 | $ | 2,875,167 | $ | - | $ | 4,020,167 | |||||||||||
| Chairman and Chief Executive Officer(3) | 2021 | $ | 615,000 | $ | 430,000 | $ | 1,290,323 | $ | - | $ | 2,335,323 | |||||||||||
| Steve O’Loughlin | 2022 | $ | 400,000 | $ | 185,000 | $ | 891,144 | $ | - | $ | 1,476,144 | |||||||||||
| Chief Financial Officer | 2021 | $ | 370,000 | $ | 150,000 | $ | 447,034 | $ | - | $ | 967,034 | |||||||||||
| (1) | The bonus disclosed in this column relates to performance in the prior year, but was contingent upon board approval, and was paid in the year disclosed. |
| (2) | The dollar amounts in this column represent the aggregate grant date fair value of all option awards granted during the indicated year. These amounts have been calculated in accordance with FASB ASC Topic 718, using the Black-Scholes option-pricing model. For a discussion of valuation assumptions, see Note 6 to our financial statements. These amounts do not necessarily correspond to the actual value that may be recognized from the option awards by the NEOs. |
| (3) | In addition to the foregoing, on August 17, 2022, Mr. Seth was granted an award of 300,000 restricted stock units, or RSUs, which were granted in exchange for warrants that Mr. Seth received for services provided to the Company prior to becoming employed by Actinium. These warrants were granted on December 17, 2012 and vested and became exercisable on the 12-month anniversary of the grant date. The warrants were in the money for their entire existence since vesting. Mr. Seth was appointed Chairman of the Board in October 2013, became Executive Chairman in August 2014 and Chief Executive Officer in June 2017. Mr. Seth refrained from exercising the warrants in order to be aligned with the long-term interests of the Company and shareholders. In November 2018, the Board extended the expiration of Mr. Seth’s warrants to February 2022. In February 2022, the Company requested that Mr. Seth not exercise the warrants to maintain alignment with the long-term interests of the Company. In exchange for refraining from exercising these warrants, the Board determined to grant Mr. Seth 300,000 RSUs based on the average fair value of the warrants during their vested life based on the Black-Scholes option-pricing model to continue to align Mr. Seth with the long-term interest of the Company and shareholders. The RSU grant was detailed on Form 4 filed with the SEC on August 19, 2022. |
Narrative Disclosure to Summary Compensation Table
For a discussion of the material terms of each named executive officer’s employment agreement or arrangement, refer to the sections above titled “Directors, Executive Officers and Corporate Governance—Chief Executive Officer Compensation” and “Directors, Executive Officers and Corporate Governance—Chief Financial Officer/Principal Financial Officer Compensation.”
On August 17, 2022, Mr. Seth was issued 300,000 restricted stock units, or RSUs, in exchange for warrants issued to him for services provided to the Company prior to being employed by Actinium. These RSUs vest at the earliest of a change of control event, the termination of the recipient’s continuous service status for any reason other than by the Company for cause and the third anniversary of the date of the grant.
78
On July 1, 2022, Mr. Seth was granted an option to purchase 827,366 shares of common stock and Mr. O’Loughlin was granted an option to purchase 256,438 shares of common stock. The options have an exercise price of $4.96 per share and expire on July 1, 2032. Pursuant to the terms of the Company’s Amended and Restated 2019 Stock Plan, 2% of the options will vest each month from the respective dates of grants until fully vested.
On September 1, 2021, Mr. Seth was granted an option to purchase 310,182 shares of common stock and Mr. O’Loughlin was granted an option to purchase 107,463 shares of common stock. The options have an exercise price of $6.07 per share and will expire on September 1, 2031. Pursuant to the terms of the Company’s Amended and Restated 2019 Stock Plan, 2% of the options will vest each month from the respective dates of grants until fully vested.
Director Compensation
The following table sets forth the compensation of our non-employee directors for the year ended December 31, 2022:
| Name | Fees Earned | Stock Awards | Option Awards (1)(2) | All Other Compensation | Total | |||||||||||||||
| Jeffrey W. Chell | $ | 51,000 | - | $ | 250,748 | - | $ | 301,748 | ||||||||||||
| David Nicholson | $ | 63,000 | - | $ | 250,748 | - | $ | 313,748 | ||||||||||||
| Ajit J. Shetty | $ | 58,500 | - | $ | 250,748 | - | $ | 309,248 | ||||||||||||
| Richard Steinhart | $ | 63,000 | - | $ | 250,748 | - | $ | 313,748 | ||||||||||||
| (1) | The dollar amounts in this column represent the aggregate grant date fair value of all option awards granted during the indicated year. These amounts have been calculated in accordance with FASB ASC Topic 718, using the Black-Scholes option-pricing model. For a discussion of valuation assumptions, see Note 6 to our financial statements. These amounts do not necessarily correspond to the actual value that may be recognized from the option awards by the Directors. |
| (2) | At December 31, 2022, the aggregate number of option awards outstanding for each director was as follows: (i) for Dr. Chell, 112,423, (ii) for Dr. Nicholson, 115,506, (iii) for Dr. Shetty, 112,173, and (iv) for Mr. Steinhart, 117,171. |
Our non-employee directors are paid an annual fee of $40,000 and receive annual option grants. Dr. Nicholson as Lead Director receives an additional annual fee of $10,000. Board committee members receive the following compensation:
| BOD Committee | Chairman | Member | ||||||
| Audit | $ | 20,000 | $ | 6,000 | ||||
| Compensation | $ | 10,000 | $ | 5,000 | ||||
| Nominating and Corporate Governance | $ | 7,500 | $ | 3,000 | ||||
79
OUTSTANDING EQUITY AWARDS AT FISCAL YEAR-END - 2022
The following table sets forth all unexercised stock options and unvested restricted stock units that have been awarded to our named executives by the Company that were outstanding as of December 31, 2022.
| Option Awards | Stock Awards | |||||||||||||||||||||||||||||||||||
| Name (a) |
Number of Securities Underlying Unexercised Options (#) (Exercisable) (b) |
Number of Securities Underlying Unexercised Options (#) (Unexercisable) (c) |
Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#)(d) |
Option Exercise Price ($) (e) |
Option Expiration Date (f) |
Number of Shares or Units of Stock That Have Not Vested (#) (g) |
Market Value of Shares or Units of Stock That Have Not Vested ($) (h) |
Equity Incentive Plan Awards: Number of Unearned Shares, Units or Other Rights That Have Not Vested (#) (i) |
Equity or Value of Other |
|||||||||||||||||||||||||||
| Sandesh Seth | 9,333 | (1) | - | - | 183.90 | 9/23/2024 | - | - | - | - | ||||||||||||||||||||||||||
| 5,000 | (1) | - | - | 107.40 | 2/15/2025 | - | - | - | - | |||||||||||||||||||||||||||
| 16,666 | (1) | - | - | 59.70 | 4/15/2026 | - | - | - | - | |||||||||||||||||||||||||||
| 24,998 | (1) | - | - | 41.70 | 3/14/2027 | - | - | - | - | |||||||||||||||||||||||||||
| 33,333 | (1) | - | - | 23.487 | 7/13/2028 | - | - | - | - | |||||||||||||||||||||||||||
| 41,000 | (2) | 9,000 | - | 6.96 | 7/12/2029 | - | - | - | - | |||||||||||||||||||||||||||
| 77,868 | (2) | 61,194 | - | 9.55 | 8/12/2030 | - | - | - | - | |||||||||||||||||||||||||||
| 93,054 | (2) | 217,128 | - | 6.07 | 9/01/2031 | - | - | - | - | |||||||||||||||||||||||||||
| 82,736 | (2) | 744,630 | - | 4.96 | 7/01/3032 | - | - | - | - | |||||||||||||||||||||||||||
| - | - | - | - | - | 300,000 | 3,195,000 | - | - | ||||||||||||||||||||||||||||
| Steve O’Loughlin | 3,333 | (1) | - | - | 53.70 | 9/28/2025 | - | - | - | - | ||||||||||||||||||||||||||
| 1,666 | (1) | - | - | 59.70 | 4/15/2026 | - | - | - | - | |||||||||||||||||||||||||||
| 3,333 | (1) | - | - | 41.70 | 3/14/2027 | - | - | - | - | |||||||||||||||||||||||||||
| 8,833 | (2) | - | - | 23.487 | 7/13/2028 | - | - | - | - | |||||||||||||||||||||||||||
| 10,933 | (2) | 2,400 | - | 6.96 | 7/12/2029 | - | - | - | - | |||||||||||||||||||||||||||
| 33,068 | (2) | 25,998 | - | 9.55 | 8/12/2030 | - | - | - | - | |||||||||||||||||||||||||||
| 32,338 | (2) | 75,225 | - | 6.07 | 9/01/2031 | - | - | - | - | |||||||||||||||||||||||||||
| 25,643 | (2) | 230,795 | - | 4.96 | 7/01/2032 | - | - | - | - | |||||||||||||||||||||||||||
| (1) | Fully vested. |
| (2) | Pursuant to the terms of the Company’s 2013 Stock Plan or 2019 Stock Plan, 2% of these options vest each month from the date of grant. |
80
Indemnification of Directors and Officers
Section 102(b)(7) of the Delaware General Corporation Law allows a corporation to provide in its certificate of incorporation that a director of the corporation will not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duty as a director, except where the directors breached the duty of loyalty, failed to act in good faith, engaged in intentional misconduct or knowingly violated a law, authorized the payment of a dividend or approved a stock repurchase in violation of Delaware corporate law or obtained an improper personal benefit. Our certificate of incorporation provides for this limitation of liability.
Section 145 of the General Corporation Law of the State of Delaware provides that a Delaware corporation may indemnify any person who was, is or is threatened to be made, party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person is or was an officer, director, employee or agent of such corporation or is or was serving at the request of such corporation as a director, officer employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his conduct was illegal. A Delaware corporation may indemnify any persons who are, or were, a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person is or was a director, officer, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit, provided such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the corporation’s best interests, provided that no indemnification is permitted without judicial approval if the officer, director, employee or agent is adjudged to be liable to the corporation. Where an officer or director is successful on the merits or otherwise in the defense of any action referred to above, the corporation must indemnify him against the expenses which such officer or directors has actually and reasonably incurred.
Section 145 further authorizes a corporation to purchase and maintain insurance on behalf of any person who is or was a director, officer, employee or agent of the corporation or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation or enterprise, against any liability asserted against him and incurred by him in any such capacity, or arising out of his status as such, whether or not the corporation would otherwise have the power to indemnify him under Section 145.
Our bylaws provide that we will indemnify our directors and officers to the fullest extent authorized by the General Corporation Law of the State of Delaware. Expenses (including attorneys’ fees) incurred by an officer or director of the Corporation in defending any civil, criminal, administrative or investigative action, suit or proceeding may be paid by the Company in advance of the final disposition of such action, suit or proceeding upon receipt of an undertaking by or on behalf of such director or officer to repay such amount if it shall ultimately be determined that such person is not entitled to be indemnified by the Company as authorized under Delaware law. Such expenses (including attorneys’ fees) incurred by former directors and officers or other employees and agents of the Company or by persons serving at the request of the Company as directors, officers, employees or agents of another corporation, partnership, joint venture, trust or other enterprise may be so paid upon such terms and conditions, if any, as the Company deems appropriate.
The indemnification rights set forth above shall not be exclusive of any other right which an indemnified person may have or hereafter acquire under any bylaw, agreement, vote of stockholders or disinterested directors or otherwise, both as to action in such person’s official capacity and as to action in another capacity while holding such office, and shall continue as to a person who has ceased to be a director, officer, employee, or agent and shall inure to the benefit of the heirs, executors, and administrators of such person.
We maintain a general liability insurance policy that covers liabilities of directors and officers of our corporation arising out of claims based on acts or omissions in their capacities as directors or officers. We have also entered into Indemnification Agreements with our executive officers and directors.
At the present time, there is no pending litigation or proceeding involving a director, officer, employee, or other agent of ours in which indemnification would be required or permitted. We are not aware of any threatened litigation or proceeding that may result in a claim for such indemnification.
81
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table shows the beneficial ownership of our Common Stock as of March 31, 2023 held by (i) each person known to us to be the beneficial owner of more than five percent (5%) of any class of our shares; (ii) each director; (iii) each Named Executive Officer; and (iv) all directors and executive officers as a group.
Beneficial ownership is determined in accordance with the rules of the SEC, and generally includes voting power and/or investment power with respect to the securities held. Shares of Common Stock subject to options and warrants currently exercisable or which may become exercisable within 60 days of March 31, 2023, are deemed outstanding and beneficially owned by the person holding such options or warrants for purposes of computing the number of shares and percentage beneficially owned by such person, but are not deemed outstanding for purposes of computing the percentage beneficially owned by any other person. Except as indicated in the footnotes to this table, the persons or entities named have sole voting and investment power with respect to all shares of our Common Stock shown as beneficially owned by them.
Unless otherwise indicated, the principal address of each of the persons below is c/o Actinium Pharmaceuticals, Inc., 275 Madison Ave, 7th floor, New York, NY 10016.
| Name of Beneficial Owner | Number of Shares of Common Stock Beneficially Owned | Percentage of Ownership(a) | ||||||
| Beneficial Owners of 5% or More of Our Common Stock | ||||||||
| Michael Bigger | 1,691,667 | (1) | 6.6 | % | ||||
| Name Executive Officers and Directors | ||||||||
| Sandesh Seth | 522,029 | (2) | 2.0 | % | ||||
| Steve O’Loughlin | 163,859 | (3) | * | |||||
| Jeffrey W. Chell, M.D. | 39,915 | (4) | * | |||||
| David Nicholson, Ph.D. | 43,581 | (5) | * | |||||
| Ajit S. Shetty, Ph.D. | 40,672 | (6) | * | |||||
| Richard I. Steinhart | 45,229 | (7) | * | |||||
| All Directors and Officers as a Group (6 persons) | 855,285 | (8) | 3.2 | % | ||||
| * | less than 1% |
| (a) | Based on 25,729,370 shares of common stock outstanding as of March 31, 2023 |
| (1) | The address of record is 2250 Red Springs Drive, Las Vegas, NV 89135. Based on the beneficial owner’s Schedule 13G filed February 6, 2023, shares beneficially owned consist of 416,000 shares of Common Stock owned by Bigger Capital Fund, LP (“Bigger Capital”), 708,167 shares of Common Stock owned by District 2 Capital Fund LP (“District 2 CF”), 172,500 shares of Common Stock held by Mr. Bigger through an IRA and another account, 135,000 shares of Common Stock through an IRA held by Patricia Winter, the spouse of Mr. Bigger, and an aggregate of 260,000 shares of Common Stock through an IRA held by the sons of Mr. Bigger. Mr. Bigger is also the beneficial owner of 66,406 shares of Common Stock issuable upon exercise of Warrants owned by Bigger Capital and 96,666 shares of Common Stock issuable upon exercise of Warrants owned by District 2 CF. The warrants are subject to a 4.99% beneficial ownership limit. The number of shares and percentage set forth above assume the no exercise of the warrants due to the beneficial ownership limit. Mr. Bigger disclaims beneficial ownership of these securities. |
| (2) | On September 23, 2014, Mr. Seth was granted an option to purchase 9,333 shares of common stock with an exercise price of $183.90 per share. On February 18, 2015, Mr. Seth was granted an option to purchase 5,000 shares of common stock with an exercise price of $107.40 per share. On April 15, 2016, Mr. Seth was granted an option to purchase 16,666 shares of common stock at an exercise price of $59.70 per share. On March 14, 2017, Mr. Seth was granted options to purchase an aggregate of 24,998 shares of common stock at an exercise price of $41.70 per share. On July 13, 2018, Mr. Seth was granted an option to purchase 33,333 shares of common stock at an exercise price of $23.487 per share. On July 12, 2019, Mr. Seth was granted an option to purchase 50,000 shares of common stock at an exercise price of $6.96 per share. On August 12, 2020, Mr. Seth was granted an option to purchase 139,062 shares of common stock at an exercise price of $9.55 per share. On September 1, 2021, Mr. Seth was granted an option to purchase 310,182 shares of common stock at an exercise price of $6.07 per share. On July 1, 2022, Mr. Seth was granted an option to purchase 827,366 shares of common stock at an exercise price of $4.96 per share. All options are subject to vesting. Within 60 days of March 31, 2023, options to purchase an aggregate of 516,648 shares of common stock will have vested. Includes 5,381 shares of common stock. |
82
| (3) | On October 1, 2015, Mr. O’Loughlin was granted options to purchase 3,333 shares of common stock with an exercise price of $53.70 per share. On April 15, 2016, Mr. O’Loughlin was granted options to purchase of 1,666 shares of common stock at an exercise price of $59.70 per share. On March 14, 2017, Mr. O’Loughlin was granted options to purchase 3,333 shares of common stock at an exercise price of $41.70 per share. On July 13, 2018, Mr. O’Loughlin was granted an option to purchase 8,833 shares of common stock at an exercise price of $23.487 per share. On July 12, 2019, Mr. O’Loughlin was granted an option to purchase 13,333 shares of common stock at an exercise price of $6.96 per share. On August 12, 2020, Mr. O’Loughlin was granted an option to purchase 59,066 shares of common stock at an exercise price of $9.55 per share. On September 1, 2021, Mr. O’Loughlin was granted an option to purchase 107,463 shares of common stock at an exercise price of $6.07 per share. On July 1, 2022, Mr. O’Loughlin was granted an option to purchase 256,438 shares of common stock at an exercise price of $4.96 per share. Within 60 days of March 31, 2023, options to purchase an aggregate of 162,676 shares of common stock will have vested. Includes 1,183 shares of common stock. |
| (4) | On April 27, 2018, Dr. Chell was granted an option to purchase 2,500 shares of common stock with an exercise price of $10.41 per share. On July 13, 2018, Dr. Chell was granted an option to purchase 2,500 shares of common stock at an exercise price of $23.487 per share. On July 12, 2019, Dr. Chell was granted an option to purchase 8,333 shares of common stock at an exercise price of $6.96 per share. On August 12, 2020, Dr. Chell was granted an option to purchase 8,333 shares of common stock at an exercise price of $9.55 per share. On September 1, 2021, Dr. Chell was granted an option to purchase 18,351 shares of common stock at an exercise price of $6.07 per share. On July 1, 2022, Dr. Chell was granted an option to purchase 72,156 shares of common stock at an exercise price of $4.96 per share. All options are subject to vesting. Within 60 days of March 31, 2023, options to purchase an aggregate of 39,915 shares of common stock will have vested. |
| (5) | On February 18, 2015, Dr. Nicholson was granted an option to purchase 833 shares of common stock with an exercise price of $107.40 per share. On April 15, 2016, Dr. Nicholson was granted an option to purchase 2,500 shares of common stock at an exercise price of $59.70 per share. On March 14, 2017, Dr. Nicholson was granted an option to purchase 2,500 shares of common stock at an exercise price of $41.70 per share. On July 13, 2018, Dr. Nicholson was granted an option to purchase 2,500 shares of common stock at an exercise price of $23.487 per share. On July 12, 2019, Dr. Nicholson was granted an option to purchase 8,333 shares of common stock at an exercise price of $6.96 per share. On August 12, 2020, Dr. Nicholson was granted an option to purchase 8,333 shares of common stock at an exercise price of $9.55 per share. On September 1, 2021, Dr. Nicholson was granted an option to purchase 18,351 shares of common stock at an exercise price of $6.07 per share. On July 1, 2022, Dr. Nicholson was granted an option to purchase 72,156 shares at an exercise price of $4.96 per share. All options are subject to vesting. Within 60 days of March 31, 2023, options to purchase an aggregate of 43,248 shares of common stock will have vested. Includes 333 shares of common stock. |
| (6) | On March 28, 2017, Dr. Shetty was granted an option to purchase 2,500 shares of common stock with an exercise price of $47.40 per share. On July 13, 2018, Dr. Shetty was granted an option to purchase 2,500 shares of common stock at an exercise price of $23.487 per share. On July 12, 2019, Dr. Shetty was granted an option to purchase 8,333 shares of common stock at an exercise price of $6.96 per share. On August 12, 2020, Dr. Shetty was granted an option to purchase 8,333 shares of common stock at an exercise price of $9.55 per share. On September 1, 2021, Dr. Shetty was granted an option to purchase 18,351 shares of common stock at an exercise price of $6.07 per share. On July 1, 2022, Dr. Shetty was granted an option to purchase 72,156 shares at an exercise price of $4.96 per share. All options are subject to vesting. Within 60 days of March 31, 2023, options to purchase an aggregate of 39,915 shares of common stock will have vested. Includes 757 shares of common stock. |
| (7) | On December 16, 2013 Mr. Steinhart was granted an option to purchase 1,665 shares of common stock at an exercise price of $201.00 per share. On February 18, 2015, Mr. Steinhart was granted an option to purchase 833 shares of common stock at an exercise price of $107.40 per share. On April 15, 2016, Mr. Steinhart was granted an option to purchase 2,500 shares of common stock at an exercise price of $59.70 per share. On March 14, 2017, Mr. Steinhart was granted an option to purchase 2,500 shares of common stock at an exercise price of $41.70 per share. On July 13, 2018, Mr. Steinhart was granted an option to purchase 2,500 shares of common stock at an exercise price of $23.487 per share. On July 12, 2019, Mr. Steinhart was granted an option to purchase 8,333 shares of common stock at an exercise price of $6.96 per share. On August 12, 2020, Mr. Steinhart was granted an option to purchase 8,333 shares of common stock at an exercise price of $9.55 per share. On September 1, 2021, Mr. Steinhart was granted an option to purchase 18,351 shares of common stock at an exercise price of $6.07 per share. On July 1, 2022, Mr. Steinhart was granted an option to purchase 72,156 shares at an exercise price of $4.96 per share. All options are subject to vesting. Within 60 days of March 31, 2023, options to purchase an aggregate of 44,913 shares of common stock will have vested. Includes 316 shares of common stock. |
| (8) | Includes options to purchase 847,315 shares of common stock and 7,970 shares of common stock. |
83
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Transactions with Related Persons
None.
Director Independence
For disclosures regarding our policies relating to director independence, refer to the section above titled “Directors, Executive Officers and Corporate Governance—Corporate Governance—Director Independence.”
Non-Competition Agreements
Our executive officers have signed non-competition agreements, which provide that all inventions become the immediate property of us and require invention assignments. The agreements provide that the executive officers will hold proprietary information in the strictest confidence and not use the confidential information for any purpose not expressly authorized by us.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The table below shows the
aggregate fees billed for professional services for the audits and audit-related fees of the Company’s annual financial statements
included in Form 10-K for the years ending December 31, 2022 and 2021, respectively, by Marcum LLP (PCAOB ID Number
| Year Ended December 31, 2022 | Year Ended December 31, 2021 | |||||||
| Audit Fees | $ | 178,793 | $ | 128,400 | ||||
| Audit – Related Fees | 29,540 | 20,600 | ||||||
| Tax Fees | - | - | ||||||
| All Other Fees | - | - | ||||||
| Total | $ | 208,333 | $ | 149,000 | ||||
Audit Fees. This category includes the audit of our annual consolidated financial statements, reviews of our financial statements included in our Form 10-K and Form 10-Qs and services that are normally provided by our independent registered public accounting firm in connection with its engagements for those years.
Audit-Related Fees. This category consists of assurance and related services by our independent registered public accounting firm that are reasonably related to the performance of the audit or review of our financial statements and are not reported above under “Audit Fees.” The services for the fees disclosed under this category include consents regarding equity issuances.
Pre-Approval Policy
In 2015, the Audit Committee adopted policies and procedures for the pre-approval of audit and non-audit services performed by the independent registered public accountants pursuant to which the Audit Committee generally is required to pre-approve the audit and permissible non-audit services performed by the independent registered public accountants in order to ensure that the provision of such services does not impair the registered accountants’ independence.
All of the services rendered by Marcum in 2022 were pre-approved by the Audit Committee.
84
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
85
86
| * | Filed herewith. |
| ** | Furnished herewith. |
| # | Indicates a management contract or compensatory plan or arrangement. |
| + | Certain of the schedules (and similar attachments) to this Exhibit have been omitted in accordance with Regulation S-K Item 601(a)(5) of Regulation S-K under the Securities Act of 1933, as amended, because they do not contain information material to an investment or voting decision and that information is not otherwise disclosed in the Exhibit or the disclosure document. The registrant hereby agrees to furnish a copy of all omitted schedules (or similar attachments) to the SEC upon its request. |
| † | Portions of this exhibit have been omitted pursuant to Item 601(b)(10)(iv) of Regulation S-K under the Securities Act of 1933, as amended, because they are both (i) not material and (ii) the type that the registrant treats as private or confidential. A copy of the omitted portions will be furnished to the SEC upon its request. |
87
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant.
| Dated: March 31, 2023 | ACTINIUM PHARMACEUTICALS, INC. | |
| By: | /s/ Sandesh Seth | |
| Sandesh Seth | ||
| Chairman and Chief Executive Officer (Duly Authorized Officer, Principal Executive Officer) | ||
| By: | /s/ Steve O’Loughlin | |
| Steve O’Loughlin | ||
| Chief Financial Officer (Duly Authorized Officer, Principal Financial and Accounting Officer) | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following person on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Sandesh Seth | Chairman and Chief Executive Officer | March 31, 2023 | ||
| Sandesh Seth | (Principal Executive Officer) | |||
| /s/ Jeffrey Chell | Director | March 31, 2023 | ||
| Jeffrey Chell | ||||
| /s/ David Nicholson | Director | March 31, 2023 | ||
| David Nicholson | ||||
| /s/ Richard I. Steinhart | Director | March 31, 2023 | ||
| Richard I. Steinhart | ||||
| /s/ Ajit J. Shetty | Director | March 31, 2023 | ||
| Ajit J. Shetty |
88